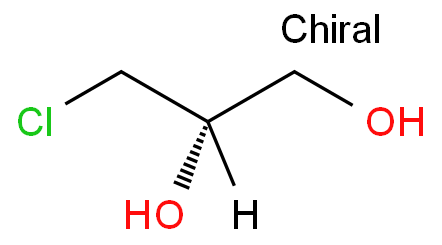
1
1
本研究旨在探讨合成并应用(S)-3-氯-1,2-丙二醇的方法,希望通过这项研究为相关领域的合成化学和应用研究提供新的思路和实验支持。
简述:3-氯-1,2-丙二醇(3-monchloropropane-1,2-diol.3-MCPD),也称alpha-chlorohydrin(α-CH),是重要的化工生产和药物合成的中间体,也作为杀菌剂使用。人工合成的3-MCPD有三种形式,即R,S和R/S混合型。
1. (S)-3-氯-1,2-丙二醇的合成:
1.1 方法一:
(1)外消旋环氧氯丙烷的拆分
在500 mL三口烧瓶中加入外消旋环氧氯丙烷350 mL (4.484 mol), [(R,R)-N,N'-双-(3,5-二叔丁基水杨基)-1,2- 环己二胺(2-)]乙酸钴2.96 g (0.00448 mol), 在室温下缓慢滴加水44 mL (2.466 mol), 反应明显放热, 滴毕在室温下继续搅拌, 气相色谱跟踪检测。 反应毕,水环真空泵蒸出 (S)-(+)-环氧氯丙烷。
(2)(S)-3-氯-1,2-丙二醇的制备
在500 mL三口烧瓶中加入配制好的2%稀硫酸120g, 缓慢升温到80~85 ℃, 滴加S-环氧氯丙烷160 mL (2.0 mol), 滴毕升温至105 ℃, 反应3 h。冷却至室温, 用30%氢氧化钠溶液中和到pH=7. 减压蒸馏, 收集130~136 ℃/2.67 kPa的馏分, 得168 g无色粘稠液体 4,产率76.5%, 气相色谱含量>99℅。
1.2 方法二:
将350 mL (4.473 mol) 外消旋环氧氯丙烷和2.97 g (0.00447 mol) (S, S) Salen Co(Ⅲ)OAc催化剂加入到500 mL 反应瓶中。在0℃冰浴条件下,缓慢滴加36.2 mL (2.01 mol) 水,滴毕后保持在0~5℃反应48小时。当反应结束(通过气相色谱检测环氧氯丙烷和氯二醇的含量比不再变化)后,进行减压蒸馏(60~62℃/13.3 kPa),得到无色透明液体(R)-环氧氯丙烷,收率为42.3%,沸点为116℃,气相色谱含量为99.0%。随后,再用油泵减压(130~132℃/2.67 kPa)蒸馏,得到(S)-3-氯-1,2-丙二醇,收率为37.2%,气相色谱含量为99.3%。
2. 应用:制备(S)-美托洛尔。
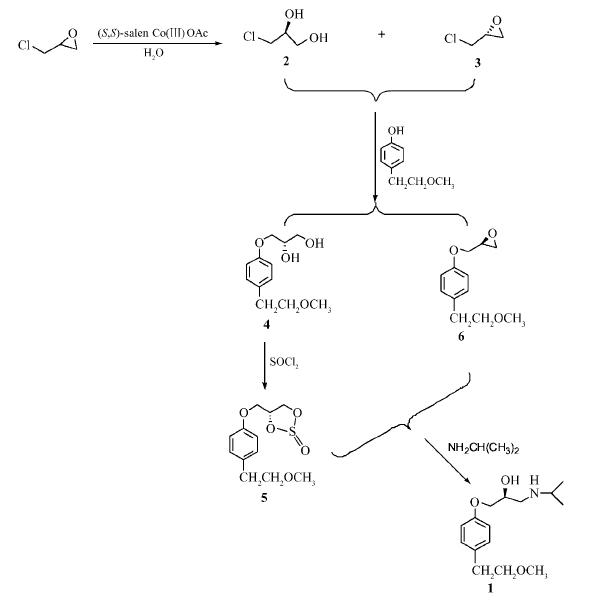
2.1 (2S)-3-[4-(2-甲氧基乙基苯氧基)-丙烷-1,2-二醇](4)的合成
在装有回流冷凝管、滴液漏斗、温度计的500 mL四口瓶中加入30.4 g(0.2 mol)4-(2-甲氧基乙基) 苯酚和250 mL无水乙醇,控制反应温度不超过5℃冰浴中分批加入8.8 g(0.22 mol)NaOH,加毕,滴加 24.3 g(0.22 mol)(S)-3-氯-1,2-丙二醇(2)和20 mL无水乙醇的混合液。滴毕,于室温反应5 h,再升温至70℃反应5 h。反应完毕,趁热过滤,滤液浓缩的粗品,加入正己烷回流至透明清亮,置冰箱中冷却,析出固体,抽滤得40 g白色针状物4,收率88.5%。
2.2 (4R)-4-[4-(2-甲氧基乙基苯氧甲基)-1,3,2-二氧硫戊环-2-氧化物](5)的合成
将22.6 g(0.1 mol)化合物4溶于100 mL二氯甲烷中,加入14 mL三乙胺,并用冰盐浴冷至-10℃,缓慢滴加17.85 g(0.15 mol)SOCl2和20 mL二氯甲烷的混合液。滴毕于-10℃继续反应4 h,向反应液中缓慢加入100 mL水,分出有机层,水层再用二氯甲烷(30 mL×3)萃取,合并二氯甲烷相,分别用水(50 mL×2)、盐水(50 mL×2)洗涤,无水硫酸钠干燥,减压蒸除溶剂得23.1 g油状物5,收率 85.0%。
2.3 (2S)-3-[4-(2-甲氧基乙基苯氧基)-1,2-环氧丙烷](6)的合成
在配有搅拌器和温度计的500 mL三口瓶中加入4-(2-甲氧基乙基)苯酚30.4 g(0.2 mol)和250 mL DMSO,冰浴中滴加8.8 g(0.22 mol)质量分数5%的NaOH溶液,控制反应温度不超过5℃,滴加55.5 g (0.6 mol)(R)-环氧氯丙烷(3),撤去冰浴,室温反应12 h后,加入100 mL蒸馏水,用二氯甲烷(50 mL× 3)萃取。合并有机层,用饱和盐水洗涤,无水硫酸钠干燥,过滤,减压蒸馏,收集122~124℃/0.66 kPa 的馏分,得32.6 g无色油状物6,收率78.4%。
2.4 (S)-美托洛尔的制备(1)
分别由中间体5和6合成。由中间体5合成:在100 mL三口瓶中加入13.6 g(0.05 mol)化合物5、18.9 g(0.32 mol)异丙胺,加热回流2 h,反应完全后蒸出过量的异丙胺,用石油醚重结晶得11.6 g白色固体化合物1,收率87.1%,mp 41~42℃;由中间体6合成:在100 mL三口瓶中加入16.6 g(0.08 mol)化合物6、18.9 g (0.32 mol)异丙胺,加热回流5 h,反应完全后蒸出溶剂和过量的异丙胺,用石油醚重结晶多次得19.0 g 白色固体化合物1,收率89.0%。
参考文献:
[1]宋光伟,朱锦桃,姚国新等. (S)-美托洛尔的不对称合成 [J]. 应用化学, 2010, 27 (11): 1286-1290.
[2]王燕,沈大冬,朱锦桃. (S)-和(R)-普萘洛尔的不对称合成 [J]. 有机化学, 2007, (05): 678-681.
[3]钱国庆,张皓,张国州等. R,S及R/S型3-氯-1,2-丙二醇的急性毒性研究 [J]. 卫生研究, 2007, (02): 137-140.
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手