4,5-二氯嘧啶-2-胺是一种常用的医药合成中间体。如果不慎吸入4,5-二氯嘧啶-2-胺,请将患者移到新鲜空气处;如果皮肤接触,应立即脱去污染的衣着,用肥皂水和清水彻底冲洗皮肤,如有不适感,应就医;如果眼睛接触,应分开眼睑,用流动清水或生理盐水冲洗,并立即就医;如果误食,应立即漱口,禁止催吐,并立即就医。
制备4,5-二氯嘧啶-2-胺的方法如下:首先向圆底烧瓶中加入2-氨基-5-氯嘧啶-4-醇和过量的POCl3,然后在110℃下反应3小时。接着加入饱和的NaHCO3水溶液以调节pH值大于8。过滤混合物,收集滤液,得到目标化合物。该方法得到的收率为18%。
1HNMR(DMSO-d6,300MHz):8.35(s,1H),7.33(s,2H)。
ESI-MS理论计算值C4H435Cl2N3[M+H]+=163.98,实验测得:164.00。
4,5-二氯嘧啶-2-胺可用作医药合成中间体。例如,可以用它来制备4-(6′-溴-1′-吲哚啉基)-5-二氯嘧啶-2-胺。具体步骤是:向含有4,5-二氯嘧啶-2-胺和6-溴吲哚啉的圆底烧瓶中加入异丙醇和浓盐酸,然后回流过夜。接着用旋转蒸发仪浓缩反应混合物,加入NaHCO3饱和水溶液以调节pH值大于8。水层用乙酸乙酯萃取,然后依次用饱和食盐水洗、无水硫酸钠干燥,最后用旋转蒸发仪浓缩。通过快速柱层析法纯化粗品,可以得到目标化合物。该方法得到的收率为40%。
1HNMR(DMSO-d6,300MHz):11.02(s,1H),8.20(s,1H),7.19(d,J=7.86Hz,1H),7.16(dd,J=7.86,1.76Hz,1H),4.12(t,J=8.40Hz,2H),3.10(t,J=8.56Hz,2H)。
ESI-MS理论计算值C12H1179Br35ClN4[M+H]+=324.99,实验测得:325.00。
[1] CN105769871 NIK蛋白激酶抑制剂作为制备治疗肝病的药物的应用
吡唑并嘧啶是一类结构特殊的含氮杂环化合物,具有广泛的生物活性。因此,近年来吡唑并嘧啶类化合物的合成及活性研究越来越引起研究者的兴趣。其中,最具有代表性的就是吡唑并[3,4-d]嘧啶化合物。基于吡唑并[3,4-d]嘧啶结构的生物活性分子研究开发取得了重要进展,例如抗肿瘤活性、抗菌活性、抗病毒活性和抗炎活性。因此,研究吡唑并[3,4-d]嘧啶化合物结构母核合成具有重要的意义和价值,并为高效的抗肿瘤药物的研发提供前期基础。
吡唑并[3,4-d]嘧啶结构的合成方法较多,可以吡唑环为母环合成吡唑并[3,4-d]嘧啶衍生物或以嘧啶环为母环合成吡唑并[3,4-d]嘧啶衍生物。本实验以2-氨基-4,6-二氯嘧啶-5-甲醛为起始物料,经与水合肼关环制备得到目标化合物4-氯-1H-吡唑并[3,4-D]嘧啶-6-胺。

图1 4-氯-1H-吡唑并[3,4-D]嘧啶-6-胺的合成反应式
准备1000ml圆底烧瓶,加入无水乙醇500ml,2-氨基-4,6-二氯嘧啶-5-甲醛,升温,搅拌下加入200ml冰醋酸、冰浴冷却,慢慢滴加水合肼,滴加完毕加热反应6h,蒸去冰醋酸,加水,用碳酸钠调pH=8,干燥,回收溶剂,得油状物140g,薄层色谱检测反应,显示单一斑点,未精制,直接用于下一步反应。将硫酸二甲酯加到上述油状物中,加热反应2.5h,加入CH2Cl280ml,Na2CO3水溶液洗涤,干燥,回收溶剂,得粗产品125g。加入NaOH的醇水溶液400ml,加热回流,搅拌反应4h,抽去乙醇,残留液加入浓HCl,析出淡黄色固体,过滤,水洗至中性,烘干,得产品4-氯-1H-吡唑并[3,4-D]嘧啶-6-胺91克。
[1] Synlett, , # 13 p. 1900 - 1904
本文将讲述2,5-二氯嘧啶作为亲电试剂与不同氮杂环胺进行偶联反应的效果,旨在为相关领域的研究人员提供参考依据。
简述:2,5-二氯嘧啶,英文名称:2,5-dichloropyrimidine,CAS:22536-67-0,分子式:C4H2Cl2N2。2,5-二氯嘧啶是一种反应物,已用于合成BI 207524,BI 是一种吲哚二酰胺NS5B Thumb Pocket 1抑制剂,用于治疗慢性丙型肝炎病毒。
亲电试剂氯代嘧啶:
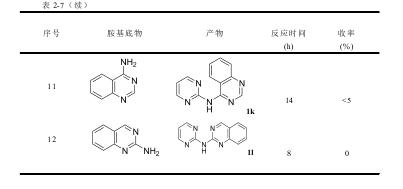
刘路显等人氯代嘧啶与氮杂环胺的Buchwald-Hartwig C-N交叉偶联反应,并在此基础上,将C-N交叉偶联反应应用于新型嘧啶类ALK小分子抑制剂的合成中。首先以2-氯嘧啶与2-氨基吡嗪的偶联反应为研究对象,对该反应进行条件优化,N-(2-吡嗪基)-2-嘧啶胺收率为84.4%。其次,为了考察适合此反应体系的胺基底物种类,研究了2-氯嘧啶与不同氮杂环胺进行偶联反应的效果,结果表明:亲核试剂氮杂环胺不同导致了收率的不同,氮杂环中氮原子的个数、苯环的引入以及氨基取代基的位置与氮杂环胺的亲核能力紧密相关,单环且氮原子个数少的氮杂环胺化合物,其对应的产物收率较高。
最后,为了进一步考察此反应体系的普遍性,使用5-氯嘧啶,2,5-二氯嘧啶与氮杂环胺进行反应,结果表明:对于亲电试剂氯代嘧啶,2,5-二氯嘧啶的亲电能力最高,2-氯嘧啶的反应活性高于5-氯嘧啶。具体如下:
(1)5-氯-N-(2-吡啶)-2-嘧啶胺(3a)的合成的具体步骤为:向100 mL反应瓶中依次加入2-氨基吡啶(94.2 mg,1.00 mmol),2,5-二氯嘧啶 (161 mg,1.40 mmol),醋酸钯[Pd(OAc)2](11.3mg,0.05 mmol),1,1'-双(二苯膦)二茂铁[DPPF](56.3 mg,0.10 mmol),碳酸铯(978 mg,3.00 mmol,3.0当量)。然后用注射器注入10 mL无水1,4-二氧六环,打开磁力搅拌,通氩气排放空气重复操作至液面无气泡生成,电加热磁力搅拌器控制实验温度,油浴加热慢缓升高温度至110℃,混合物在此温度下搅拌2 h。反应结束冷却至室温,加入40 mL水和60 mL 乙酸乙酯分离各相,水相用更多的乙酸乙酯(3×100 mL)萃取,合并有机相,用无水硫酸钠干燥,过滤并真空浓缩。将粗产物用溶剂梯度(石油醚/乙酸乙酯=3:1,2:1,1:1,v:v)进行硅胶柱色谱分离,得到目标产物5-氯-N-(2-吡啶)-2-嘧啶胺 190 mg,白灰色结晶,收率为92%。
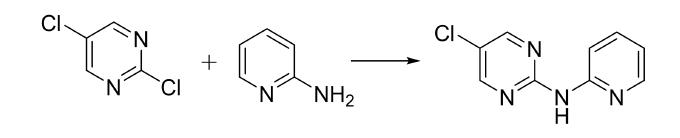
(2)表2-9为2,5-二氯嘧啶与氮杂环胺的实验结果。对比亲电试剂2,5-二氯嘧啶 与2-氯嘧啶,表2-9中1-4与表2-7中1-2,10,12,可以发现2,5-二氯嘧啶底物 的收率较高,5-氯-N-(2-吡啶)-2-嘧啶胺(3a)5-氯-N-(2-吡嗪)-2-嘧啶胺(3b)、N-(5-氯嘧啶-2-基)喹喔啉-2-胺(3c)、N-(5-氯嘧啶-2-基)喹唑啉-2-胺(3d)的收率分别为92%、86%、71%、43%,对应的2-氯嘧啶底物的收率降低甚至不反应,N-(2-吡啶基)-2-嘧啶胺(1a),N-(2-吡嗪基)-2-嘧啶胺(1b)、N-(2-嘧啶基)-喹喔啉-2-胺(1j)、N-(2-嘧啶基)-喹唑啉-2-胺(1l)的收率分别为90%、84%、34%、0。结果表明,2,5-二氯嘧啶的反应活性高于5-氯嘧啶。当卤代杂芳烃上连接吸电基团时,其亲电能力增强。2,5-二氯嘧啶相比2-氯嘧啶,其对位上连接了一个氯吸电基团,进一步提高了2,5-二氯嘧啶的亲电能力,故其对应的产物收率较高些。



参考文献:
[1]刘路显.Buchwald-Hartwig C-N偶联反应用于嘧啶衍生物及新型ALK小分子抑制剂的合成研究[D].天津大学,2019.DOI:10.27356/d.cnki.gtjdu.2019.002294
N-甲基-N-((3R,4R)-4-甲基哌啶-3-基)-7H-吡咯并[2,3-D]嘧啶-4-胺,是一种白色至类白色固体,具有显著的碱性,难溶于水但是可溶于酸性水溶液和强极性有机溶剂。这种化合物可用作有机合成中间体和医药化学基础试剂,在医药领域主要用于生物活性分子托法替尼的制备。
N-甲基-N-((3R,4R)-4-甲基哌啶-3-基)-7H-吡咯并[2,3-D]嘧啶-4-胺结构中含有多个氮原子,表现出显著的碱性,可与多种酸性物质结合成盐。此外,它还可进行亲核取代反应和缩合反应,用于制备衍生物。
![N-甲基-N-((3R,4R)-4-甲基哌啶-3-基)-7H-吡咯并[2,3-D]嘧啶-4-胺的酰胺化反应](https://imgen4.guidechem.com/img/answer/2024/10/15/1728993668523727.jpg "N-甲基-N-((3R,4R)-4-甲基哌啶-3-基)-7H-吡咯并[2,3-D]嘧啶-4-胺的酰胺化反应")
图1 N-甲基-N-((3R,4R)-4-甲基哌啶-3-基)-7H-吡咯并[2,3-D]嘧啶-4-胺的酰胺化反应
在一个干燥的反应烧瓶中将N-甲基-N-((3R,4R)-4-甲基哌啶-3-基)-7H-吡咯并[2,3-D]嘧啶-4-胺和其他试剂混合,可以得到目标产物分子。
N-甲基-N-((3R,4R)-4-甲基哌啶-3-基)-7H-吡咯并[2,3-D]嘧啶-4-胺主要用于生物活性分子托法替尼的制备。托法替尼是一种JAK抑制剂,对多种炎症相关疾病有良好的治疗效果。
[1] Gehringer, Matthias; et al ChemMedChem,2014,9,2516-2527.
2-氯-8-环戊基-5-甲基-8H-吡啶并[2,3-D]嘧啶基-7-酮是一种杂环芳烃类化合物,可用作有机合成中间体。它的英文名为2-chloro-8-cyclopentyl-5-Methylpyrido[2,3-d]pyriMidin-7(8H)-one,化学式为C13H14ClN3O,CAS号为1013916-37-4,分子量为263.72。该化合物的熔点为176度到178度,密度为1.3,外观为黄色或暗黄色固体。
2-氯-8-环戊基-5-甲基-8H-吡啶并[2,3-D]嘧啶基-7-酮在有机合成中具有广泛的应用。它可以进行偶联反应,得到烷基化和芳基化的产物。此外,通过溴化反应,可以在双键上靠近羰基的位置引入一个溴原子。
然而,需要注意的是,2-氯-8-环戊基-5-甲基-8H-吡啶并[2,3-D]嘧啶基-7-酮作为一种含卤的有机化合物,对水环境具有较大的危害。因此,在使用过程中应避免未稀释或大量产品接触地下水、水道或污水系统。
为了保持其稳定性,2-氯-8-环戊基-5-甲基-8H-吡啶并[2,3-D]嘧啶基-7-酮应密封储存于低温(最好在2到8度)和干燥的环境中,最好是惰性气体保护的贮藏器内。同时,应避免与氧化物接触,并尽量避开碱性物质。
针对2-氯-8-环戊基-5-甲基-8H-吡啶并[2,3-D]嘧啶基-7-酮的合成,常规的方法是从5-溴-2-氯-N-环戊胺嘧啶-4胺出发,经过钯催化剂的作用,发生相应的Heck反应。得到的中间产物是一个酸,该酸在酸酐的作用下进行分子内的胺酯交换反应,形成内酰胺化合物,从而得到目标产物。因此,该反应实际上是两步反应。

[1] Duan S, Place D, Perfect H H, et al. Palbociclib commercial manufacturing process development. Part I: control of regioselectivity in a grignard-mediated SNAr coupling[J]. Organic Process Research & Development, 2016, 20(7): 1191-1202.
尿嘧啶(如图1)是常见且天然存在的嘧啶衍生物。最早发现于1900年,由酵母核素(nuclein,即为核酸)水解分离而得,可存在于牛的胸腺和脾脏、腓鱼的精液和小麦芽中。尿嘧啶为平面、不饱和的化合物,可吸收光线。

图1 尿嘧啶
性质
尿嘧啶存在于RNA中,与腺嘌呤形成硷基对,在DNA转录反应中,取代胸嘧啶。尿嘧啶甲基化后,产生胸嘧啶,以保护DNA,并提高DNA複製之效率。尿嘧啶以氢键与腺嘌呤配对,因为它含有氧和氮等电负度大的原子,是氢键接受者,而且可形成两个氢键。尿嘧啶也可以与核糖结合,形成核糖核苷──尿苷(如图2)。若有一个磷酸根连接在尿苷上,就形成尿苷5’-单磷酸。

图2 尿苷的化学结构
合成
在2009出版的某学术文件中,NASA的科学家报告了某种合成尿嘧啶的方法:在与太空相似的条件下,使嘧啶曝晒紫外光,可製造尿嘧啶。此研究显示最早自然发生的尿嘧啶之可能来源。
尿嘧啶有许多实用的实验室合成法。最简单的合成法就是把水与胞嘧啶混合,可以产生尿嘧啶与氨。
C4H5N3O + H2O → C4H4N2O2 + NH3
合成尿嘧啶最普遍的方法就是顺丁烯二酸与尿素,在发烟硫酸中进行缩合反应。
C4H4O4 + CH4N2O → C4H4N2O2 + 2 H2O + CO
硫尿嘧啶在氯乙酸水溶液中进行複分解,也是合成尿嘧啶的方法之一。此外,先用β-丙胺酸与尿素反应,可製造5,6-二氢尿嘧啶(5,6-diuracil,尿密啶的5,6两个碳原子各多接了一个氢原子);再让5,6-二氢尿嘧啶进行光脱氢反应,可合成尿嘧啶(如图3)。
?
图3 利用β-丙胺酸与尿素合成尿嘧啶
反应
尿嘧啶很容易进行一些普通的反应,包括氧化、硝化及烷基化。当有PhOH/NaOCl存在时,在UV光的蓝光区,可以看见尿嘧啶。因为有一个以上提供电子的原子团,尿嘧啶也有能力与元素态卤素反应。
尿嘧啶很容易与核糖、磷酸根进行加成反应,以便参与体内的合成及进一步反应。尿嘧啶变成尿苷、尿苷单磷酸(UMP)、尿苷二磷酸(UDP)、尿苷三磷酸(UTP)及尿苷二磷酸葡萄糖(UDP-葡萄糖)。上述每种分子都在体内合成,并各自具有专一的功能。
尿嘧啶与核糖、磷酸根键结可协助合成许多细胞运作必需的酶,这就是尿嘧啶在体内的用途。 尿嘧啶可作为异位(allosteric)调节剂,以及人体与植物中化学反应的辅酶。UMP控制植物的胺甲醯磷酸合成酶(carbamoyl phosphate synthetase)和天冬胺酸转胺甲醯酶,而UDP及UTP调节动物的CPSaseⅡ(胺甲醯磷酸合成酶Ⅱ)。UDP-葡萄糖利用醣代谢反应将肝脏及其他组织中的葡萄糖转化成半乳糖。尿嘧啶也参与多醣类的生物合成与醛糖的运输。
用途
尿嘧啶可以用于药物传递(drug delivery)及作为药剂。元素态的氟与尿嘧啶反应,生成5-氟尿嘧啶。5-氟尿嘧啶是抗癌药(抗代谢药),在複製胺基酸的过程中,5-氟尿嘧啶可冒充尿嘧啶。因为5-氟尿嘧啶的形状与尿嘧啶很相似,但化学性质不同,这种药可以抑制RNA複製酶,因而减缓RNA合成,并使癌细胞的成长停止。
参考资料:
1. http://en.wikipedia.org/wiki/Uracil
2. http://en.wikipedia.org/wiki/Drug_delivery
3. http://en.wikipedia.org/wiki/Carbamoyl_phosphate_synthetase
4. http://uracil.navajo.cz/
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手




























