天然活性化合物在新药研发中具有重要作用,肉桂酸及其酯类衍生物是一类常见的天然化合物,存在于多种植物中。这些化合物具有抗癌、抗病毒、抗微生物等活性,其中(E)-肉桂酸酯类化合物尤为引人关注。然而,目前尚未有关于(E)-肉桂酸酯类化合物抗植物病原菌活性的研究报道。
对香豆酸乙酯的制备方法如下:将乙酸酯膦叶立德和相应芳香醛在无水乙醇中反应,经过回流搅拌后,蒸除溶剂得到沉淀。沉淀经过柱层析分离,洗脱后得到目标化合物对香豆酸乙酯。
该化合物为白色棒状晶体,产率为78%,熔点为74~75℃。其1HNMR和ESI-MS的谱图也得到了确认。
[1] CN201610934356.1(E)-肉桂酸酯类化合物及其合成方法、以及含有该化合物的药物及应用
对羟基肉桂酸(p-Hydroxycinnamic acid),又称对香豆酸,是医药与香料工业中间体,广泛存在于自然界植物中。除了通过有机合成获得,还可以直接从天然产物中提取纯化获得。

可以采用90%乙醇提取,大孔吸附树脂纯化,40%乙醇洗脱,浓缩干燥,乙酸乙酯再回流提取的方法来得到对香豆酸粗品。
通过碱解、超滤、透过液脱色、离子交换吸附、解吸、二次脱色、结晶等步骤对玉米芯中主要成分对香豆酸进行含量富集。
蔗渣先经碱解,滤液超滤,活性炭脱色,离子交换树脂吸附,浓缩结晶即可得到高含量对香豆酸。
以上几种方法大多需要使用柱层析方式纯化对香豆酸,该工艺周期较长,回收率偏低,而且溶剂消耗较大。
CN105384626B公开了一种新的分离纯化竹茹中对香豆酸的方法,步骤如下:
步骤1:向原料竹茹中加入原料质量3倍量的水和原料质量0.3%-0.8%的复合生物酶,得酶解后原料;所述复合生物酶为木质过氧化物酶和木葡聚糖酶的混合酶;所述木质过氧化物酶和木葡聚糖酶的质量比为1:1.2;
步骤2:将酶解后原料晾干或烘干,加入有机物回流提取,合并提取液并浓缩,放置,过滤,收集沉淀;所述有机物为乙酸乙酯、乙酸甲酯和正丁醇中的任意一种或多种的混合;
步骤3:步骤2所得的沉淀先用石油醚洗涤,再用酸性溶液洗涤;所述酸性溶液为盐酸、硫酸或柠檬酸的水溶液;
步骤4:向步骤3洗涤后的沉淀中加入乙醇回流溶解,然后加入活性炭吸附除杂,过滤,放置冷却析晶,干燥即得对香豆酸。
此方法的优点包括:
1.生物酶解:采用酶解-提取-洗涤-重结晶等手段对竹茹中的对香豆酸进行分离纯化,工艺简单,效果好;主要采用生物酶解的手段使对香豆酸完全的游离出来,操作条件缓和,且酶的专一性可以避免一些其他成分的分解,或遇酸碱而溶出,降低了后续处理的难度。
2.周期短、成本低:采用四步即可将竹茹中的对香豆酸进行分离纯化,首先酶解为后续的分离纯化做了很好的铺垫,再通过小极性有机溶剂提取明显减少了杂质的含量,从而避免了背景技术中各种柱层析的使用,缩短了工艺流程,适用于工业化生产。
3.质量稳定:本发明工艺简单,可操作性强,产品收率高,质量稳定,重复性高。
香豆素是一种重要的绿色荧光染料,具有较大的摩尔消光系数和量子产率。近年来,经过修饰后的香豆素被广泛应用于发光材料、化学传感器以及标记生物分子。其中,3-乙酰基羟基香豆素作为香豆素衍生物中的重要代表,具有价格低廉和灵敏度高等优点。
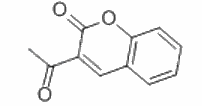
3-乙酰基羟基香豆素的结构
3-乙酰基羟基香豆素可以通过一定的制备方法得到。首先将300mg(1.47mmol)的3-乙酰基羟基香豆素溶于15mL二氯甲烷中,然后加入508mg(3mmol)硫代氯甲酸苯酯,常温搅拌4小时。通过旋蒸过柱,可以得到粗产品。如果需要纯品,可以将其过柱,然后在二氯甲烷和石油醚的混合体系中进行重结晶。最终可以得到白色纯净的3-乙酰基羟基香豆素,产率为42%。
该探针对汞离子有很好的响应,随着探针溶液中Hg2+浓度的增加,荧光强度逐渐增强。在(0-2μM)Hg2+浓度范围内,Hg2+的浓度与荧光强度呈线性关系。因此,该探针能够实现对汞离子的灵敏识别,可以较精确地确定待测样本中汞离子的含量。
3-乙酰基羟基香豆素可以与半胱氨酸发生迈克加成环化反应,从而实现荧光探针的制备。首先,将2,4-二羟基苯甲醛与乙酰乙酸乙酯反应得到3-乙酰基羟基香豆素,然后与丙烯酰氯反应得到2-(3-乙酰基)香豆素基丙烯酸酯。
制备方法:将2,4-二羟基苯甲醛(1,20mmol),乙酰乙酸乙酯(24mmol)和1mL哌啶加入到30mL无水乙醇中,加热至回流,反应3小时后冷至室温,有绿色固体析出,抽滤,乙醇洗三次,粗产品用乙醇重结晶得到淡绿色固体2,熔点:237℃,产率91%。
在37℃条件下,将1μM的2-(3-乙酰基)香豆素基丙烯酸酯分别加入到不同浓度的牛血清溶液中培养20分钟。通过荧光酶标仪检测荧光强度,结果显示2-(3-乙酰基)香豆素基丙烯酸酯可以成功地应用于生物液体中,且血清浓度越大,荧光越强。
[1] 邓正江, & 林红梅. (2005). 3-乙酰基-5-羟甲基-7-羟基香豆素治疗病毒性肝炎高胆红素血症37例. 蚌埠医学院学报, 30(6), 553-553.
[2] 蒋德炉, 朱广军, 邓汝温, & 吴集贵. (1995). 3—乙酰乙酰基—4—羟基香豆素希土配合物的合成和性质. 无机化学学报, 11(1), 15-18.
[3] 张珍英, 邓慧敏, & 邓芹英. (2008). 香豆素类新基质用于基质辅助激光解吸/电离飞行时间质谱对聚乙二醇的测定. 分析测试学报, 27(11), 1225-1228.
探讨如何合成7 - 二乙氨基 - 4 - 甲基香豆素不仅有助于深入了解其合成途径,还可以为其在材料科学等领域的应用提供重要参考。
简述:7 - 二乙氨基 - 4 - 甲基香豆素(SWN),是一种香豆素型的荧光增白剂。香豆素本身就具有非常强烈的荧光,在其3和7位上引入取代基团就可使其成为具有实用价值的荧光增白剂,其白度高、耐光性能优良、增白后的制品不带蓝光头或红光头,制品外观悦目,在洗涤行业中应用广泛,SWN 即为此类的代表之一。
合成:
1. 方法一:
以原料二乙替间氨基酚和β-丁酮酸乙酯在复合催化剂存在下,制得7-二乙氨基-4-甲基香豆素。经过实验得到合适的工艺路线,即二乙替间氨基酚和β-丁酮酸乙酯在金属锌粉、四氯化钛和乙酸酐的存在下,加热进行成环缩合反应,有效缩短反应时间,提高产物收率和纯度,减少废水的污染。具体步骤如下:

在配有回流冷凝管及分水器的干燥的四口烧瓶中,加入二乙替间氨基酚165 g(1 mol),β-丁酮酸乙酯195 g(1.5 mol),油浴升温至60℃,分别加入 6.0 g金属锌粉、四氯化钛21 g和乙酸酐10.2 g。于 75~80℃保温2 h,继续加热至反应物回流,并收集挥发液。在温度110℃反应4 h,直至无挥发液产生。反应物冷却后(<50℃),立即加入盛有65 g石油醚溶液的容器内,夹套冷却至10℃以下,析出固体,经精制、低温减压干燥得197.5 g 7-二乙氨基-4-甲基香豆素,以二乙替间氨基苯酚计,产物的收率为85.6%。产物的熔点范围为72.4~73.2℃。
2. 方法二:
(1)将间二乙氨基苯酚16.5克(0.1克分子)和氯化锌、氯化亚锡(二者的量的关系及与其它反应物的量的关系)混合后加入烧瓶中,然后加入26ml(0.2克分子)的乙酰乙酸乙酯在油浴上加热回流,反应3.5小时,冷却至室温;
(2)将上述反应后的混合物放入浓度为12%稀硫酸中,析出带紫色的粒状结晶体,滤出结晶产物,在 40℃下干燥,得到7-二乙氨基 -4-甲基香豆素粗产物;
(3)将7-二乙氨基-4-甲基香豆素粗产品用乙醇重结晶2次,然后再再用石油醚重结晶2次,即得到满足激光使用要求的白色针状结晶的激光染料7-二乙氨基 -4-甲基香豆素。
该方法通过对激光染料7-二乙氨基-4-甲基香豆素粗产品进行酸洗,多次重结晶的工艺处理后,可有效地去除激光染料7-二乙氨基-4-甲基香豆素粗产品中所含有的杂质,有效地提高激光染料7-二乙氨基-4-甲基香豆素的纯度,延长其在使用中的寿命,显著提高激光染料7-二乙氨基-4-甲基香豆素制备的经济性。
参考文献:
[1]王磊,杨晋青,顾宇翔. 高效液相色谱-三重四级杆串联质谱法测定化妆品中的7-二乙氨基-4-甲基香豆素 [J]. 香料香精化妆品, 2018, (04): 35-39.
[2]黄胜. 7-二乙氨基-4-甲基香豆素合成中催化剂的改良 [J]. 上海化工, 2009, 34 (06): 16-18. DOI:10.16759/j.cnki.issn.1004-017x.2009.06.004.
[3]天津市化学试剂研究所. 激光染料7-二乙氨基-4-甲基香豆素的制备方法. 2010-05-26.
对羟基肉桂酸(P-Hydroxycinnamic acid)又称对香豆酸。广泛存在于自然界植物中,尤其以豆科植物含量居多。对羟基肉桂酸广泛应用于医药、食品、日用品、饲料等化工领域。本文将介绍一种以对羟基苯甲醛为主要原料制备对羟基肉桂酸的方法[1].

以往的对羟基肉桂酸制备工艺主要有以下几种:
① 天然提取法难度大、成本高,且产率极低;
② 以往的合成工艺中采用对羟基苯甲醛与丙二酸为原料,按照Knoevenagel-doebner反应合成了对羟基肉桂酸,但收率低,收率仅为50%;
③ 以对羟基苯甲醛和丙二酸、吡啶、哌啶在微波条件下进行反应,收率虽有提高,但是原材料的成本相应提高,实现工业化生产较为复杂。
(1)在反应容器中加入乙酸乙酯700ml,对羟基苯甲醛97.6g,搅拌,降温;
(2)当温度降到10-30℃时开始加入甲醇钠150g,控制温度20-50℃,加入甲醇钠后搅拌,在20—50℃之间保温;
(3)保温结束后,加入水200-500ml,充分搅拌,开始蒸馏,直至蒸馏到99℃;
(4)蒸馏结束后降温到50℃以下,慢慢加入20%的液碱600ml,再加热升温到回流,保温;保温结束后降温至60℃,开始慢慢滴加30%的盐酸,调pH=1,降温到40℃以下,过滤,洗涤,抽干、干燥;得对羟基肉桂酸成品。
此方法以廉价的乙酸乙酯为原料,来源广泛,易于采购;以价廉易购的甲醇钠为催化剂,能够降低成本;原料成本仅为常规生产工艺的2/3;合成的收率高;反应的时间短;使用的溶剂沸点低,容易蒸馏回收利用,降低能耗,从而降低生产成本。
[1] 一种对羟基肉桂酸的制备工艺. CN102351689A
己酸烯丙酯是一种无色至微黄色透明液体,具有强烈的菠萝样的果香香气,并有朗姆酒似的风味,天然品存在于菠萝等中。菠萝醛被FEMA认定为GRAS,FEMA编号2032,并经FDA批准食用,我国GB2760-1996规定为暂时允许使用的食用香料,广泛用于调配菠萝、苹果、杏、桃子、甜橙、草莓等果香型香精,以及老姆酒和白酒等酒用香精。

一种己酸烯丙酯的生产工艺,其特征在于:包括以下步骤:
A、酯化:酯化釜按比例投入己酸、烯丙醇和对甲苯磺酸,重量比为:2-甲基丁酸:乙醇:对甲苯磺酸=55:10:1,全回流1hr后塔顶出料,等顶温升至92℃,同时自高位槽进入2-甲基丁酸150L/hr、乙醇80L/hr,控制酯化釜蒸汽压力0.5Mpa;
B、减压分馏:酯化釜蒸出的粗酯经油水分离器进入粗蒸釜进行减压蒸馏,控制真空度在-0.08Mpa,蒸汽压力0.3Mpa左右,釜温不低于120℃,顶温控制在50-60℃,塔顶蒸出的低沸物(烯丙醇)泵入酯化釜;
C、减压精馏:釜低料通过泵经流量计进入精馏釜,控制精馏釜真空度-0.09Mpa,顶温控制在110-115℃,塔顶蒸出的成品通过泵经流量计进入己酸烯丙酯成品槽,精馏釜釜底高沸物(己酸)通过泵回入酯化釜。
在配有温度计、分水器、回流冷凝管、电动搅拌器的四颈瓶中加入一定量的丙烯醇,己酸(0.1 mol),对苯二酚(0.05 g),甲苯以及催化剂(纳米稀土复合超强酸La3+/SO42- /TiO2),启动搅拌器,控制温度,回流反应一定时间后,静置,冷却,倾出反应液。分出有机相,合并后,进行洗涤,干燥。滤液加入氯化亚铜蒸馏(0.05g氯化亚铜/0.1 mol 菠萝醛粗品),收集186~188℃的馏分,即得产品。
应用一、CN201810814434.3公开了一种饲料用香精及其配制方法,该产品属于兽用香精制造领域,生产该产品使用的原料包括:香豆素10%-12%、丁二酮8%-10%、丁酸5%、香草醛7%、乙酸乙酯5%、肉桂醛6%、己酸烯丙酯3%、己醇6%、乙基香兰素5%、菜子油43%,其工艺是:将配方量的菜子油送入容器,边搅拌边加热至70-75℃,向容器中依次加入配方量的香豆素、丁二酮为主香剂,丁酸、香草醛、乙酸乙酯、肉桂醛、己酸烯丙酯、己醇,搅拌均匀后反应25-27分钟;停止加热,降温至常温后,继续搅拌,加入配方量的乙基香兰素,搅拌12-14分钟后停止搅拌得到成品。
应用二、CN201310589083.8公开了一种食品乳化剂,以重量份计,本发明由以下组分组成:甘油单脂肪酸酯:4~6,失水山梨醇单硬脂酸酯:3~5,山梨醇:10~20,柠檬酸钠:1~3,丁酸香叶酯:10~20,己酸烯丙酯:10~20,甘油:20~30。与现有技术相比,本发明具有如下有益效果:(1)乳化效果好,用途广泛、原料易得、成本低廉;(2)可用于糕点、奶油、果糖等的乳化。
[1]CN201310433722.1一种己酸烯丙酯的生产工艺
[2]CN201810814434.3一种饲料用香精
[3]CN201310589083.8一种食品乳化剂
[4]张福捐,盛淑玲.纳米稀土复合超强酸催化合成菠萝醛[J].食品科技,2009,34(01):225-227.
2-溴苯乙酮是一种有机中间体,可通过苯乙酮与液溴反应制得。它在有机合成中具有重要的应用价值,特别是在生产医药中间体、氮茚衍生物、香豆素衍生物和香料等方面。

将600mmol(72.0g)苯乙酮溶解于500mL二氯甲烷中,然后在0℃下滴入2mol液溴(320g),搅拌3小时。反应结束后,通过减压蒸馏除去溶剂,得到2-溴苯乙酮。
2-溴苯乙酮可用于制备N-苯甲酰甲基溴吡啶。制备过程包括苯乙酮与液溴在-20-40℃下反应,反应时间为2~8小时。得到的2-溴苯乙酮与吡啶在一定溶剂中在0-80℃下反应3-15小时,然后通过减压除去溶剂,加水和乙酸乙酯分液,用水洗涤有机相,再经过无水硫酸钠干燥。最后,通过减压蒸馏除去溶剂,得到纯品。
一种基于柱前衍生的LC-MS测定有机酸含量的方法被公开。该方法通过将待测样品、碳酸钠和2-溴苯乙酮加入溶剂中,在50~60℃下进行衍生化反应20~28小时。然后,取反应产物进行液相色谱-质谱分析,通过对照标准曲线计算待测样品中有机酸的含量。该方法通过化学修饰技术对有机酸进行衍生化处理,提高离子化效率,降低基质干扰,同时减少无机盐及内源性杂质的干扰,改善被分析物的信号强度。通过液相色谱-质谱联用技术分析检测衍生化产物,能实现对有机酸的定性和定量分析,具有简单、高效、检测灵敏度高和准确性好等优点。
[1] [中国发明] CN201611037583.0 一种N-苯甲酰甲基溴吡啶的制备方法
[2] CN201610277903.3基于柱前衍生的LC-MS测定有机酸含量的方法
保泰松是一种属于吡唑酮类衍生物的药物,主要作用于中枢神经系统。它具有较强的抗炎作用,对炎性疼痛有良好的效果,并能促进尿酸的排泄。

保泰松的解热镇痛作用较弱,一般不作为解热镇痛药使用。然而,它具有较强的抗炎作用,可用于治疗风湿性及类风湿性关节炎、急性痛风性关节炎、强直性脊椎炎等疾病。此外,保泰松还能减少尿酸盐的再吸收,对急性痛风患者有一定的排尿作用。它也可用于治疗急性血吸虫病、丝虫急性淋巴管炎、结核病、恶性肿瘤等引起的发热。
近年的研究发现,保泰松产生消炎、镇痛和抗风湿作用的机理可能是通过抑制炎症组织中产生炎症相关活性物质的合成,如前列腺素、白细胞的活动和转移、溶酶体酶的释放及其活性等。
保泰松口服后吸收迅速而完全,大约2小时后达到血药浓度峰值。它在血浆中的蛋白结合率为98%。保泰松的表观分布容积为0.12L/kg,随着剂量的增加,分布容积也会增大,但血浓度不会增加。因此,当重复使用时,其稳态血浓度不会呈线性增加。保泰松的半衰期为56~86小时。它可以通过胎盘进入胎儿体内,也可以通过乳汁排出。保泰松在肝脏中代谢,代谢产物为羟基保泰松和γ-羟基保泰松,这些代谢产物仍然具有活性,最终通过尿液排出,少量通过胆汁排出。
保泰松的副作用发生率约为10%~20%,短程使用可以减少这一比率。它对胃肠道有较大的刺激作用,可能会出现恶心、呕吐、腹痛、便秘等症状,过量使用可能导致消化道溃疡和便血。此外,保泰松还可能对其他系统产生损害,如皮疹、眩晕、血尿、肝炎等。它还可以抑制骨髓引起粒细胞减少,甚至导致再生障碍性贫血,如果停药及时,多数情况下可以恢复,因此应该定期检查血象,如果1周内没有改善,就不应再使用。保泰松与双香豆素类抗凝血药、磺胺类药物、甲磺丁脲降糖药合用时,血药浓度会增加,毒性也会增大。保泰松还具有钠、氯潴留作用,因此高血压、水肿、心力衰竭的患者禁用,并且在用药期间需要限制食盐摄入量。对于有肝、肾损害、药物过敏史、溃疡病、骨质疏松症的患者,禁止使用或者谨慎使用。
保泰松可以通过氢化偶氮苯与丁基丙二酸二乙酯进行环合反应得到。首先将氢化偶氮苯、丁基丙二酸二乙酯、甲醇钠和亚硫酸钠一起加热,剧烈回流1.5小时,生成保泰松钠盐,然后用乙酸进行酸化反应,最终得到保泰松。
通过合成2-氰甲基苯并咪唑并探讨其应用,期望为2-氰甲基苯并咪唑的研发提供有益信息。
背景:2-氰甲基苯并咪唑是一种咪唑,吡唑等衍生物,外观和形状为棕色颗粒或粉末,常用于合成染料、颜料等。
合成:
1. 方法一:在100mL三口烧瓶中加入邻苯二胺5.5g,氰乙酸乙酯6.0g,对甲苯磺酸0.2g,邻二氯苯40ml,通氮气保护,升温至164~165℃,反应10h,降至室温,过滤,用少量邻二氯苯洗至无溶剂,烘干,得产品8.3g。
2. 方法二:将2.16g (0.02mol)邻苯二胺和3.20mL(0.03mol)氰基乙酸乙酯于50mL烧瓶中混合均匀,将混合物在 185~195℃油浴中加热反应2h,然后倒入干燥的烧杯中,冷却2h,抽滤,滤饼经重结晶得2-氰甲基苯并咪唑,熔点;209~211℃。
应用:
1. 合成荧光黄GX染料,具体步骤如下:
(1)3-(2-苯并咪唑基)-7-二乙氨基香豆素亚胺的合成
在100mL三口烧瓶中加入4-N、N-二乙氨基-2-羟基苯甲醛3.0g,2-氰甲基苯并咪唑2.5g,哌啶0.2g和 DMF20mL,在25℃下搅拌反应24h,过滤,用少量无水乙醇洗涤,干燥后得产物3.2g。
(2)色基82的合成
在100mL三口烧瓶中,加入3-(2-苯并咪唑基)- 7-二乙氨基香豆素亚胺2.0g,30ml浓盐酸,升温至98~ 100℃回流5h,冷却到0~5℃后用20%氢氧化钠水溶液调节pH值至7~7.5,过滤,用水洗涤至滤液为中性,干 燥后得产物1.9g。
(3)色基82的磺化
在500mL三口瓶中加入20%发烟硫酸71.0g,搅拌下分批加入30g色基-82,控制温度60℃以下,加完料后观察物料情况,慢慢升温到120℃,反应5h,测终点。
(3)稀释
1000mL烧杯中,加400g冰水,搅拌下将上述料稀释到冰水中,温度5~10℃,过滤。滤饼用1500~2000g水洗涤(刚果红试纸测洗涤液为微蓝或不变色为终点。变色范围为3.5到5.2,碱态为红色,酸态为蓝紫色)尽量抽干。滤饼重约为130g。
(4)转型
1000mL三口瓶中,加入上述湿物料和100g水打浆,搅拌下滴加液碱(40%NaOH)11g,调pH 8~9,升温到90℃,复测pH值,直至物料全溶。趁热过滤,滤渣用少量80~90℃热水洗涤,合并滤液。
(5)盐析
上述滤液加入到1000mL三口烧瓶中,加热到58~ 62℃,加液体体积的10%精盐。染料析出后,继续搅拌 1h,染料呈颗粒状后再降温到35~40℃。点样看渗圈情况再补加盐。过滤,滤瓶用10%的100mL精盐水洗涤一并过滤。湿滤饼在85℃的烘干箱中烘干。即得产品。
2. (2E,2E’)-3,3’-苯基-1,3-联苯并咪唑丙烯腈(1)和(E)-2-(2-苯并咪唑)-3-(3-吲哚)丙烯腈(2)的合成
将157mg(1.0mmol)的 2-氰甲基苯并咪唑与醛(间苯二甲醛0.5mmol或吲哚甲醛1.0mmol)溶于15mL无水乙醇中,加入1滴吗啡啉和1滴冰乙酸,将反应混合物加热回流,用TLC监测反应,直至反应完成。将反应冷至室温,把析出的固体过滤,用少量乙醇洗涤,干燥后分别得到目标化合物1和2。
参考文献:
[1]赵龙;李刚. 荧光黄GX染料的合成 [J]. 辽宁丝绸, 2022, (04): 19-20+25.
[2]杨鹏辉;孙伟. 新型苯并咪唑取代的丙烯腈及丙烯酸酯衍生物的催化合成 [J]. 化学通报, 2015, 78 (12): 1170-1172. DOI:10.14159/j.cnki.0441-3776.2015.12.022
本文将探讨合成盐酸普罗帕酮的不同方法和路线,以期为该药物的工业生产提供可行的合成方案。通过对不同合成方法的比较分析,我们旨在为盐酸普罗帕酮的合成提供更加经济高效的解决方案。
背景:盐酸普罗帕酮是一种安全、有效、并且广谱的抗心律失常药,当其他抗心律失常药无效时,对部分易感性病人可采用盐酸普罗帕酮控制心律失常。随着临床上使用的增加,盐酸普罗帕酮的作用和效果得到了进一步的肯定。自1979年在德国首次上市以来,至今仍作为抗心律失常的临床用药,然而,其合成路线报道并不多。
合成:
1. 路线一:王文洲等研究报告指出,以氯苄为起始原料,通过与四丁基溴化铵(TBAB)和碳酸钾催化的反应制备苄基丙二酸二乙酯。随后,经过与苯酚反应得到3-苄基-4-羟基香豆素,接着在碱性条件下生成2’-羟基二氢查尔酮,并与环氧氯丙烷反应生成2’-(2,3-环氧丙氧基)-3-苯基苯丙酮,最终与正丙胺反应得普罗帕酮,随后盐酸酸化得盐酸普罗帕酮。该合成路线共包含七个步骤反应,其中合成3-苄基-4-羟基香豆素需要280-290℃高温反应,对设备要求较高;而在合成2’-(2,3-环氧丙氧基)-3-苯基苯丙酮时,需使用甲醇钠甲醇溶液碱化,再蒸除甲醇,操作相对复杂,因此有必要简化操作并优化合成路线。

路线二:李秀珍等的研究指出,通过苯酚与乙酸酐酯化,再经过Fries重排反应得到邻羟基苯乙酮。在碱性条件下,并利用溴化四丁铵作为催化剂,邻羟基苯乙酮与苯甲醛反应,形成2’-羟基查尔酮,随后通过氢气还原得到2’-羟基二氢查尔酮,接着与环氧氯丙烷反应生成2’-(2,3-环氧丙氧基)-3-苯基苯丙酮,最终与正丙胺反应得到普罗帕酮,随后盐酸酸化得盐酸普罗帕酮。该合成路线的原料易得,但重排反应会产生对位异构体,导致分离困难和收率较低,此外,氢化反应需要使用昂贵的催化剂,对技术设备要求较高,操作繁琐。

路线三:史卫明等研究提出了一种合成盐酸普罗帕酮的简便路线,该方法以2’-羟基-3-苯基苯丙酮和环氧氯丙烷为原料,在四丁基溴化铵催化、碱性条件下进行反应制备2’-(2,3-环氧丙氧基)-3-苯基苯丙酮,随后蒸馏后,加入正丙胺胺化,再经盐酸酸化,即可得到盐酸普罗帕酮。虽然这种路线操作简便,适合工业生产,但其中所需的原料2’-羟基-3-苯基苯丙酮需要定制,并且价格较贵,导致成本上升。

戴长浩等人以路线三为基础,同时对2’-羟基-3-苯基苯丙酮进行合成,从而降低成本,设计以下合成路线:以邻羟基苯乙酮为起始原料,与苯甲醛在碱性条件下缩合,得到2’-羟基查尔酮,钯炭催化氢化,生成2’-羟基-3-苯基苯丙酮,环氧氯丙烷为溶剂,同时作为反应物与2’-羟基-3-苯基苯丙酮反应,生成2’-(2,3-环氧丙氧基)-3-苯基苯丙酮,蒸除回收剩余环氧氯丙烷,加入正丙胺,反应得普罗帕酮,加入18%盐酸酸化,得盐酸普罗帕酮。

参考文献:
[1]王文洲,曹胜利,潘长敏.盐酸普罗帕酮的合成[J].中国医药工业杂志,1992,(05):
[2]戴长浩.Synthesis Study for Flunarizine Hydrochloride and Propafenone Hydrochloride[D].山东大学,2019.
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手
























