香柑醇是一种从植物羌活中提取的活性成分。羌活是一种伞形科植物,主要产于四川、青海、甘肃等地。羌活具有散寒、祛风、除湿、止痛的功效,主要用于治疗风寒感冒、风湿痹痛等疾病。羌活中的化学成分主要包括挥发油、呋喃香豆素类、植物甾醇、低聚糖、有机酸、氨基酸等。
香柑醇可以用于制备具有维生素C转运体产生促进作用的物质。这种物质可以通过提取日本厚朴木兰、杏仁、甘茶、葡萄柚等植物的提取物来获得。
香柑醇可以通过特定的提取方法获得,具体方法请参考下图:

通过高效液相色谱-串联质谱(HPLC-MS/MS)法可以同时测定枳壳厚朴汤冻干粉中异欧前胡素、佛手柑内酯、花椒毒酚、橙皮油素、6',7'-羟基佛手柑素、佛手柑素以及香柑醇等7个香豆素类成分的含量。该方法具有快速、灵敏、准确的特点。
[1] 来源:天然药物化学成分提取分离手册
[2] CN200780005481.3维生素C转运体产生促进剂
[3] HPLC-MS/MS法同时测定枳壳厚朴汤冻干粉中7个香豆素类成分的含量
爱维莫潘作为一种重要的药物,其合成一直是化学领域中备受关注的课题。
简述:爱维莫潘为一种新型的外周 μ 型阿片受体拮抗剂,临床上用于防治手术以及使用阿片类药物导致的胃肠功能紊乱,特发性便秘以及肠易激综合征等,具有疗效确切、不良反应小的优势,其在手术的应用中具有良好的前景。
合成:
1. 方法一:
3-[(3R,4R)-3,4- 二甲基哌啶 -4- 基] 苯酚 (2) 与 2- 苄基丙烯酸乙酯发生 Michael 加成反应,然后通氯化氢气体析晶得(S)-2- 苄基 -3-[(3R,4R)-4-(3- 羟基苯基 )-3,4- 二甲基哌啶-1-基 ] 丙酸乙酯盐酸盐 (3),析晶母液经消旋化循环套用,3 的收率为 60%。3 依次经水解、甘氨酸异丁酯缩合、水解制得爱维莫潘,总收率约45%( 以 2 计 )。具体实验步骤如下:
(1)(S)-2- 苄基 -3-[(3R,4R)-4-(3- 羟基苯基 )-3,4-二甲基哌啶 -1- 基 ] 丙酸乙酯盐酸盐 (3)
将 2(纯度 99.4%,2.0 g,9.74 mmol)、2- 苄基丙烯酸乙酯 (2.04 g,10.72 mmol) 和甲苯(10 ml) 加至 50 ml 反应瓶中,升温回流反应 4 h。冷却至室温,加入乙醇 (14 ml),通氯化氢气体调至 pH 1,搅拌 15 min,浓缩至干。向残留物中加入乙醇 (20 ml),缓慢析出白色固体,搅拌过夜,0 ℃搅拌 1.25 h,过滤,滤饼干燥后加入乙醇 ( 约 5.2 ml)回流打浆 2 h,冷却至室温,0 ℃搅拌 1 h,过滤,干燥得白色固体 3(1.4 g,34% )。乙醇母液浓缩至干,加入乙酸乙酯 (15 ml)和水 (15 ml),加饱和碳酸钠溶液调至 pH 9 ~ 10,室温搅拌 1 h。分取有机相,用水 (10 ml) 洗涤,经水硫酸钠干燥后过滤,滤液浓缩,残留物中加入甲苯 (2.4 ml),升温回流搅拌 8 h。冷却至室温,加入乙醇 (8.5 ml),通氯化氢气体调至 pH 1,搅拌15 min,浓缩至干,加入乙醇 (12 ml) 缓慢析出白色固体,室温搅拌 2 h,0 ℃搅拌 1.25 h,过滤,滤饼干燥后在乙醇 (2.5 ml) 中回流打浆,得白色固体3(0.72 g)。收集母液,再次重复上述游离、消旋、析晶操作,与前面所得 3 合并,共制得 3 2.47 g,收率 60%。
(2)(S)-2- 苄基 -3-[(3R,4R)-4-(3- 羟基苯基 )-3,4-二甲基哌啶 -1- 基 ] 丙酸一水合物 (4)
将 3(7.65 g,17.71 mmol)、水 (70 ml) 和 50%氢氧化钠溶液 (6.1 g) 加至 250 ml 反应瓶中,室温搅拌 10 h。TLC[ 展开剂 :二氯甲烷∶甲醇 (8 ∶ 1)]显示反应完全,过滤,滤液中加入甲醇 (73.4 ml),加浓盐酸 ( 约 10.0 g) 调至 pH 6,析出固体。浓缩除去甲醇,室温搅拌 4 h,加浓盐酸重新调至 pH 6,0 ℃搅拌 1.5 h,过滤,滤饼干燥得白色固体 4(6.65 g,97.4% ),mp 183.3 ~ 185.7 ℃。
(3)2-[[(2S)-2-[[(3R,4R)-4-(3- 羟基苯基 )-3,4- 二甲基哌啶 -1- 基 ] 甲基 ]-3- 苯基丙酰 ] 氨基 ] 乙酸异丁酯 (5)
氮气保护下,将 4(6.65 g,17.2 mmol)、甘氨酸异丁酯对甲苯磺酸盐 (5.91 g,19.5 mmol) 和 1- 羟基苯并三唑 (HOBt,2.63 g,19.5 mmol) 加至 THF(67 ml) 中,控制温度不高于 15 ℃,加入三乙胺(1.97 g,19.5 mmol) 和 EDCI(3.74 g,19.5 mmol),25 ℃反应 4 h。TLC[ 展开剂 :二氯甲烷∶甲醇(8 ∶ 1)] 显示反应完全,加入乙酸乙酯 (83 ml)稀释,依次用 0.5 mol/L 的碳酸钠 - 碳酸氢钠溶液(pH ≈ 10,83 ml,称取碳酸钠 19.08 g、碳酸氢钠 26.88 g,加水定容至 1 L 即得 )、饱和氯化钠溶液 (83 ml) 和水 (83 ml) 洗涤,减压浓缩,得浅黄色油状液体 5(8.54 g,100% )。
(4)爱维莫潘 (1)
将 5(8.29 g,17.25 mmol) 加入乙醇 (209 ml)和水 (49 ml) 的混合溶剂中,控制温度小于 25 ℃,加入 1 mol/L 氢氧化钠溶液 (51.7 ml),25 ℃搅拌45 min,TLC[ 展开剂 :二氯甲烷∶甲醇 (5 ∶1)]显示反应完全。加浓盐酸调至 pH 6,析出固体,浓缩除去乙醇,室温搅拌 2 h,0 ℃搅拌 1.5 h,过滤,干燥,所得 1 粗品 (6.93 g) 经乙醇∶水 (2∶1) 重结晶,得白色固体粉末 1(6.2 g,78% ),mp207.6~208.7 ℃。纯度 99.9%。
2. 方法二:
(1)3-[(3R,4R)-3,4- 二甲基哌啶 -4- 基 ] 苯酚(4)的合成
3- 溴苯酚 (5) 在碳酸钾作用下与 2- 溴丙烷反应得到异丙基 -3- 溴苯醚 (6),6 先与镁发生格氏反应,后与1,3-二甲基哌啶-4-酮反应得到(3S,4R)-1,3-二甲基 -4-[3-(1- 甲基乙氧基 ) 苯基 ]-4- 哌啶醇(7),7 与氯甲酸乙酯反应,经对甲基二苯甲酰酒石酸 [(+)-DTTA] 拆分生成碳酸 -(3S,4R)-1,3- 二甲基 -4-[3-(1- 甲基乙氧基 ) 苯基 ]-4- 哌啶乙酯(8),8 在十氢萘作用下加热得到 (3R)-1,3- 二甲基 -4-[3-(1- 甲基乙氧基 ) 苯基 ]-1,2,3,6- 四氢吡啶(9),9 在正丁基锂作用下与硫酸二甲酯发生甲基化反应得到(3R)-1,3,4-三甲基-4-[3-(1-甲基乙氧基)-苯基 ]-1,2,3,4- 四氢吡啶 (10),10 先经硼氢化钠还原,后经 (+)-DTTA 拆分得到 (3R,4R)-1,3,4- 三甲基 -4-[3-(1- 甲基乙氧基 ) 苯基 ] 哌啶 (11),11 与氯甲酸苯酯反应得到 (3R,4R)-3,4- 二甲基 -4-[3-(1-甲基乙氧基 ) 苯基 ] 哌啶甲酸苯酯 (12),12 经酸水解得到 4 。
(2)爱维莫潘的合成
3-[(3R,4R)-3,4- 二甲基哌啶 -4- 基 ] 苯酚(4)与丙烯酸甲酯 (30) 加成得到 (3R,4R)-4-(3-羟基苯基 )-3,4- 二甲基 -1- 哌啶丙酸甲酯 (31),31先在二异丙基氨基锂 (LDA) 作用下,与苄基溴反应,再经盐酸酸化得到 (2S)-2-[[(3R,4R)-4-(3-羟基苯基 )-3,4- 二甲基哌啶 -1- 基 ] 甲基 ]-3- 苯基丙酸甲酯盐酸盐 (32),32 经水解得到 (2S)-2-[[(3R,4R)-4-(3-羟基苯基)-3,4-二甲基哌啶-1-基]-甲基 ]-3- 苯基丙酸一水合物 (33) [2] 。33 经三乙胺、DCC、1- 羟基苯并三唑 (HOBt) 作用与甘氨酸乙酯盐酸盐 (34) 反应得到 2-[[(2S)-2-[[(3R,4R)-4-(3-羟基苯基 )-3,4- 二甲基哌啶 -1- 基 ] 甲基 ]-3- 苯基丙酰 ] 氨基 ] 乙酸乙酯一水合物 (35),35 经水解得到爱维莫潘。
参考文献:
[1]王家会,赵传猛,张福利. 爱维莫潘的合成 [J]. 中国医药工业杂志, 2015, 46 (08): 799-802. DOI:10.16522/j.cnki.cjph.2015.08.001.
[2]石权达,闫冬,高静等. 爱维莫潘关键中间体的合成 [J]. 中南药学, 2015, 13 (05): 490-493.
[3]王家会,包如胜,裘鹏程等. 爱维莫潘合成路线图解 [J]. 中国医药工业杂志, 2014, 45 (03): 291-294. DOI:10.16522/j.cnki.cjph.2014.03.025.
TRANSGLUTAMINASE转谷氨酰胺酶1抗体是一种多克隆抗体,能够特异性结合转谷氨酰胺酶1。该抗体主要用于多种免疫学实验,包括Western Blot、IHC-P、IF、ELISA、Co-IP等。
谷氨酰胺转氨酶是一种分子量约38000的单体蛋白质,由331个氨基酸组成。它能够催化蛋白质多肽发生共价交联,从而改善蛋白质的结构和功能。

TGs是一组Ca2+结合及Ca2+赖性酶,其中包括TG1、TG2、TG3和TG5。这些TGs在人类表达,并在不同的组织中发挥不同的功能。TG1、TG3和TG5在表皮表达,参与角质包膜的形成。TG2在表皮和真皮中表达,参与胶原纤维及其他细胞外基质的形成。
人的TG1表达在多种复层上皮中,但酶活性仅限于颗粒层与角质层交界处。TG1的表达受维A酸的调节。
转谷氨酰胺酶是一种通过催化蛋白质之间发生共价交联反应的转移酶。茂源链霉菌的转谷氨酰胺酶酶原基因经过PCR扩增后,通过密码子优化和连接到表达载体,成功在大肠杆菌中表达。研究结果表明,密码子优化和定点突变技术可以优化转谷氨酰胺酶的表达。
为了解决转谷氨酰胺酶酶原降解的问题,研究人员在酶原和成熟肽之间引入了特异性识别位点,成功地激活了转谷氨酰胺酶。这项研究为转谷氨酰胺酶的研究和应用提供了新的思路。
[1]Investigations on the activation of recombinant microbial pro-transglutaminase:in contrast to proteinase K,dispase removes the histidine-tag[J].Christian Sommer,Thomas C.Hertel,Christian E.H.Schmelzer,Markus Pietzsch.Amino Acids.2012(2)
[2]Model based optimization of the fed-batch production of a highly active transglutaminase variant in Escherichia coli[J].Christian Sommer,Norbert Volk,Markus Pietzsch.Protein Expression and Purification.2010(1)
[3]Over-production of various secretory-form proteins in Streptomyces lividans[J].Shuhei Noda,Yuko Ito,Nobuaki Shimizu,Tsutomu Tanaka,Chiaki Ogino,Akihiko Kondo.Protein Expression and Purification.2010(2)
[4]Enzymatic properties of transglutaminase produced by a new strain of Bacillus circulans BL32 and its action over food proteins[J].LWT-Food Science and Technology.2010(2)
[5]王坤.茂源链霉菌转谷氨酰胺酶的异源表达研究[D].华南理工大学,2013.
2-叔丁基苯酚,又名邻叔丁基苯酚,是一种有机化合物,化学式为C10H14O。主要用作抗氧剂,植物保护剂,合成树脂、医药、农药中间体以及香精香料的原料。

苯酚与异丁烯发生烷基化反应,通常使用酸性物质做催化剂,由于酸催化剂的催化特性,可能会使烷基化反应过程中发生副反应,导致烷基化反应收率较低。此外,副反应 产生的重组分可能有结焦等问题,进而导致投资及运行成本较高。
李广琼[1]提供了一种制备2?叔丁基苯酚的方法,通过添加副反应抑制剂,有效抑制副反应的发生,反应收率高,且反应液不存在结焦等问题。将苯酚与异丁烯在酸性条件下进行烷基化反应,所述烷基化反应中加入2?异丙基苯酚。在苯酚的烷基化反应中,2?异丙基苯酚对该烷基化反应具有重要的促进作用,可明显提高2?叔丁基苯酚的选择性,并抑制生成焦油副反应的发生。
具体步骤为:分别向配有四斜叶浆式搅拌器的1.5升釜式不锈钢容器反应器中加入300g 苯酚、 0.35g马来酸、45g异丁烯,1g 2?异丙基苯酚和350g对特辛基酚,通过控制异丁烯进料速度,控制反应液在釜内停留时间为30min。通过氮封控制反应器压力为220kPaA,并将反应器温度控制在160℃,开启搅拌,通过换热保持反应器温度稳定在160℃。在反应过程中的不同的时间点取出反应混合物的样品,通过色谱法测量反应混合物各组分的浓度(以反应后混合物总质量计)。
通过向苯酚与异丁烯发生的烷基化反应中添加副反应抑制剂,可有效抑制副反应发生,不仅可提高烷基化反应2?叔丁基苯酚的选择性,还可促进烷基化反应正向进行,提高苯酚的转化率。优选的方案中,本发明中还通过加入稀释剂,提高了反应的热稳定性,其配合抑制剂的添加可有效减少重组分的生成,提高经济性。
[1]李广琼,闫维佳,姜庆梅等.一种制备2-叔丁基苯酚的方法[P].山东省:CN117362159A,2024-01-09.
合成3-噻吩丙二酸是有机化学领域中的重要课题,它作为一种含硫杂环化合物,具有广泛的应用价值。本文旨在探讨合成3-噻吩丙二酸的方法,为相关研究提供理论和实践上的指导。
背景:3-噻吩丙二酸(TMA)作为重要的侧链中间体与6-氨基青霉烷酸(6-APA)反应,可以制备青霉素类β-内酰胺抗生素替卡西林(ticarcillin),替卡西林与克拉维酸钾以15∶1组合形成的药物-泰门汀(timentin)是英国Smithkline-Beecham公司推出的第二个克拉维酸复方制剂。泰门汀于1986年在美国首次上市,用于治疗严重感染。
合成:
1. 方法一:
利用铜锂试剂在酯基存在下选择性发生亲核取代反应的特征,将溴代丙二酸二乙酯引入噻吩环的3-位,可合成3-噻吩丙二酸。合成路线如下:
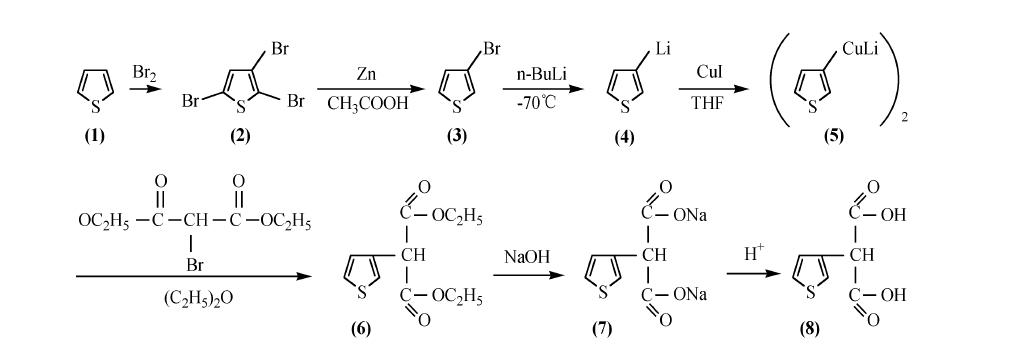
2. 方法二:
以四氢呋喃为溶剂, 丙二酸二乙酯与氢化钠反应得到丙二酸二乙酯钠, 在溴化亚铜催化作用下与3-溴噻吩反应后进行酸化生成3-噻吩丙二酸二乙酯, 其经水解酸化后得到3-噻吩丙二酸。具体步骤如下:
在150mL的三口圆底烧瓶中加入6.05g (0.2mol) 氢化钠和60mL精制过的四氢呋喃, 得到灰色悬浊液, 搅拌均匀, 缓慢升温至60℃, 之后缓慢加入精制过的丙二酸二乙酯32.85g (0.2mol) (约3h加完) 。滴毕, 继续搅拌至无气体放出, 得到丙二酸二乙酯钠。升温至回流, 向反应混合液中加入2.87g (0.02mol) 溴化亚铜及16.2g (0.1mol) 3-溴噻吩, 约6h后停止反应, 冷却至室温。搅拌下将12.44g (0.2mol) 冰醋酸加入混合物中, 生成大量固体。抽滤后得到的滤饼用20mL二氯甲烷洗涤3次, 合并有机层。得到的有机相经饱和食盐水萃取, 无水硫酸镁干燥过夜后经减压蒸馏蒸出低沸点的溶剂。在剩余液体中加入乙醇和氢氧化钠的混合水溶液, 搅拌1.5h。用活性炭脱色后蒸去其中的乙醇和水, 再用甲基异丁基酮进行萃取。在水层缓慢加入浓盐酸, 调节pH=6左右。用甲基异丁基酮再次萃取, 水相放在冰水冷却至0℃左右, 继续酸化至pH约为1。最后加入10mL乙醇, 析出固体, 过滤后将固体在40℃下真空干燥, 得到淡黄色固体。
3. 方法三:
用3-溴代噻吩对3-噻吩丙二酸进行合成,具体步骤如下:
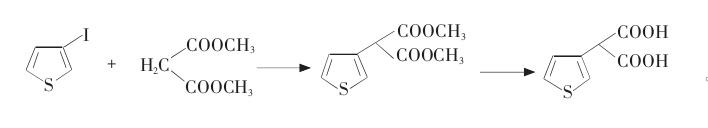
(1)3-碘代噻吩的制备
向反应瓶中加入32.58 g(0.2 mol)3-溴代噻吩、喹啉(135 mL)和CuI(58.24 g),迅速升温至140 ℃,N2保护下搅拌20 h。将反应混合物冷却至70 ℃,倒入含有135 mL冰水和135 mL盐酸的容器中,过滤出铜盐。用1,2-二氯甲烷萃取3次,分离出有机相。将有机相用质量分数为10%的NaHCO3溶液冲洗,再分离出有机相,浓缩得到3-碘代噻吩28.67 g,产率为68.3%。
(2)3-噻吩丙二酸二乙酯的制备及水解
向反应瓶中加入丙二酸二乙酯(24.03 g,0.15 mol),搅拌下加入NaH(7.2 g,质量分数为50%),反应完毕加入喹啉150 mL和CuI(23.3 g,0.12mol),在N2保护下升温至100 ℃,滴加3-碘代噻吩(20.99 g,0.1 mol),搅拌4 h。冷却至70 ℃,倒入含有150 mL冰水和150 mL浓盐酸的容器中, 过滤出铜盐,用乙酸乙酯萃取3次,合并有机相,并用质量分数为10%的NaHCO3溶液冲洗。浓缩得到3-噻吩丙二酸二乙酯20.01 g,产率为82.7%。
在烧瓶中加入2.90 g水和2.90 g(0.052 mol)氢氧化钾,搅拌溶解后,加入4.84 g(0.02 mol)3-噻吩丙二酸二乙酯,升温至回流,保持回流1~1.5 h。冷却至室温,加5.80 g水稀释,再用20 mL乙醚分2次洗涤,乙醚相回收。水层则用盐酸酸化至pH值为1,然后用30 mL乙醚分3次萃取。将有机相合并后, 滤液真空减压蒸馏回收醚后,可得浅黄色的固体,熔点为133~136 ℃。经20 mL苯重结晶、真空干燥,得熔点为138~139 ℃的白色晶体3.17 g, 收率为85.1%,总收率为48.1%。3-噻吩丙二酸的熔点为138~139 ℃。
参考文献:
[1]朱红岩,周赫元,刘颖颖等. 水溶性聚噻吩类衍生物聚(3-噻吩丙二酸)的酶催化合成与表征 [J]. 化工新型材料, 2017, 45 (05): 146-148.
[2]杨英梅,苏代国. 3-噻吩丙二酸的合成研究 [J]. 河北工业科技, 2012, 29 (06): 381-384.
[3]董运勤. 药物中间体3-噻吩丙二酸的合成研究 [J]. 中国抗生素杂志, 2008, 33 (12): 758-760.
三聚氰酸是一种三嗪类有机化合物,化学式为(CNOH)3。它是一种白色无味的固体,常被用作漂白水、杀菌剂和除草剂的成分或生产原料。全球产量超过1.6亿千克。
除了三聚氰酸,它还有其他一些名称,如氰尿酸、异氰脲酸、异氰尿酸等。
三聚氰酸是白色结晶,无气味,微苦味。它微溶于冷水,溶于热水、热醇、吡啶、浓硫酸、盐酸、氢氧化钾和氢氧化钠水溶液,不溶于醚、苯、氯仿和丙酮。
三聚氰酸是氰酸的环状三聚体,有多个互变异构体,主要以三酮形式存在。

三聚氰酸最早由弗里德里希·维勒于1829年通过尿素和尿酸的热分解制得。目前工业上主要通过尿素的聚合制取。

从水中析出的三聚氰酸含有结晶水,在空气中会失去结晶水。也可以从浓盐酸和硫酸中析出无水结晶。
三聚氰酸被广泛添加到动物饲料中,可被反刍类动物消化。它也用于合成氯代衍生物、树脂、涂料、农药除草剂等,并用于药物卤三羟嗪的生产。
三聚氰酸本身基本无毒,但在存在三聚氰胺的情况下,两者会形成不溶于水的氰尿酸三聚氰胺,可能导致肾脏衰竭。

当人体摄入三聚氰胺时,三聚氰胺中的氰尿酸三聚氰胺会被胃酸解离,导致复合物被破坏,三聚氰胺和三聚氰酸分别被吸收到血液中。由于人体无法转化这两种物质,它们在肾脏中形成固体,导致肾小管的物理阻塞,最终导致肾脏衰竭。
1H-吡咯并[2,3-B]吡啶-5-醇,也被称为5-羟基-7-氮杂吲哚,是一种常用的医药中间体。它可以用于合成ABT-199(Venetoclax),一种实验性的B细胞淋巴瘤因子-2(BCL-2)抑制剂,由艾伯维公司和罗氏公司联合开发。ABT-199旨在选择性抑制BCL-2因子的功能,恢复细胞的通讯系统,让癌细胞自我毁灭,以达到治疗肿瘤的效果。

在室温下,将30克(0.15摩尔)的5-溴-7-氮杂吲哚和25.6克(0.23摩尔)的叔丁醇钾加入1升的三口瓶中,加入400毫升四氢呋喃并搅拌溶解。然后在冰盐浴中降温至0℃,滴加80毫升稀释在80毫升四氢呋喃中的三异丙基氯硅烷(30.8克,0.16摩尔)。反应结束后,加入300毫升水并搅拌15分钟,分液。将水相用甲基叔丁基醚(200毫升*2)萃取,合并有机相。用饱和氯化钠水溶液洗涤有机相,然后用无水硫酸钠干燥,浓缩溶剂得到化合物Ⅱ。产量为49.5克,收率为92.0%。
在室温下,将4.08克(11.5毫摩尔)的化合物Ⅱ、0.6克(2.3毫摩尔)的乙酰丙酮酸铜、0.76克(2.3毫摩尔)的BHMPO、1.7克(40毫摩尔)的一水氢氧化锂、18毫升DMSO和4.5毫升水加入50毫升的反应瓶中,搅拌并用氮气置换。升温至130℃反应1小时后,停止加热并降温至室温。加入100毫升水,用2N稀盐酸调节pH值至6,有固体析出。将固体过滤,并用乙酸乙酯(50毫升*3)萃取水相。合并有机相,并用水和饱和氯化钠水溶液洗涤一次。用无水硫酸钠干燥,浓缩溶剂得到1H-吡咯并[2,3-B]吡啶-5-醇。产量为1.1克,收率为72%。
[1][中国发明]CN201710854276.X一种5-羟基-7-氮杂吲哚的制备方法
盐酸屈他维林是一种重要的化学物质,具有广泛的应用领域。对盐酸屈他维林的合成研究不仅可以深入了解其制备方法和性质特点,还有助于提高其在工业生产和科学研究中的应用效果。
简介:盐酸屈他维林(drotaverine hydrochloride,1), 化学名为1-(3,4-二乙氧基苄基)-6,7-二乙氧基- 3,4-二氢异喹啉盐酸盐,是罂粟碱类药物,对消化性溃疡、胆绞痛、急性胰腺炎、胃痉挛、肠易激综合征、阑尾炎和尿路结石等引起的腹痛都有明显的解痉止疼和缓解症状的作用。二十世纪60年代国外开始将盐酸屈他维林应用于临床,90年代开始在中国推广,目前在匈牙利、俄罗斯、中国以及其他获得批准上市的国家中广泛使用。关于盐酸屈他维林晶型的文献报道较少,仅在专利US412615A报道了盐酸屈他维林在无水乙醇中结晶成盐。在研究盐酸屈他维林精制方法及结晶条件时发现:其具有多晶型现象,在乙醇中结晶得到一种淡黄色晶体,在水中结晶得到淡黄色和类白色两种晶体,通过差示扫描量热法、粉末X-a行射法分析发现这是3种不同的晶体,故将在乙醇结晶中获得的晶体称为晶型I,在水中获得两种晶体分别称为晶型II、III。
合成:
1. 现有1的工业合成方法是从3,4-二羟基苯乙 腈(2)出发,经双乙基化反应生成3,4-二乙氧基 苯乙腈(3),3分别在Pd/C作用下催化氢化生成 3,4-二乙氧基苯乙胺(4)、在碱性条件下水解生成 3,4-二乙氧基苯乙酸(5)。用4和5经高温缩合得到 N-(3,4-二乙氧基苯乙基)-2-(3,4-二乙氧基苯基)- 乙酰胺(6)。6在三氯乙烯中经三氯氧磷催化环合生成1(图1),总收率约34%。该法虽然步骤较少,但制备起始原料2时要用到剧毒的氰化物,环境污染较大,且存在一定操作风险;制备4时需高温高压操作。

2. 有专利以图2所示路线制得1。以4-溴-1,2- 苯二酚(7)为原料,与碘乙烷经乙基化反应得到4- 溴-1,2-二乙氧基苯(8)。8与双(频哪醇合)二硼发生取代反应所得2-(3,4-二乙氧基苯基)-4,4,5,5- 四甲基-1,3,2-二氧硼杂环戊烷(9),经过Suzuki Miyaura偶联反应制得3,4-二乙氧基苯乙酸乙酯 (10)。10经氨解制得3,4-二乙氧基苯乙酰胺(11), 再经还原制得4;另用10直接水解制得5。4和5 按上法操作制得1,总收率为32%。该法虽然避免了使用剧毒化合物且收率与上述工业化方法相当,但是乙基化反应用到的碘乙烷以及Suzuki-Miyaura偶联反应中用到的催化剂1,1'-双二苯基膦二茂铁二氯化钯[Pd(dppf)Cl2]价格昂贵,生产成本相对较高。

3. 4和5是合成1的关键中间体,其高效制备是工艺路线的重点。有研究设计了一条新的合成路线(见图3),先使盐酸多巴胺(12)经(Boc)2O保护得N-Boc-多巴胺(13),13经硫酸二乙酯醚化制得 (3,4-二乙氧基苯乙基)氨基甲酸叔丁酯(14),最后水解得到4。另外参照3,4-二甲氧基苯乙酸的合成方法,用邻苯二酚(15)经硫酸二乙酯醚化得 1,2-二乙氧基苯(16),16经傅-克酰基化反应制得 3,4-二乙氧基苯乙酮酸乙酯(17),17在KOH/水 合肼作用下经黄鸣龙反应得5。5经酰氯化后再与4 经缩合、环合得到1。该路线起始原料易得,整个反应过程无需用到氰化物,生产过程和后处理都更加安全环保。制备4时直接引入氨基,无需氢气还原氰基,避免了高压反应以及氢气易燃易爆的风险。 优化后的工艺更易操作,反应条件温和,环境污染减少,总收率约60.4%(以12计),具有一定的工业化应用前景。

参考文献:
[1]王凯,阎旭冉,陈冰涵等.盐酸屈他维林的合成工艺改进[J].中国医药工业杂志,2022,53(02):192-195.DOI:10.16522/j.cnki.cjph.2022.02.004.
[2]应一锋,曾建华,叶云生.盐酸屈他维林的多晶型研究[J].华西药学杂志,2020,35(04):365-367.DOI:10.13375/j.cnki.wcjps.2020.04.004.
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手


























