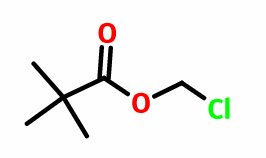
特戊酸氯甲酯是一种常用的医药中间体和化学品,具有无色透明的液体性质。然而,使用时需要注意其风险性,必须远离火源,并穿戴适当的防护服、手套和护目镜等。特戊酸氯甲酯并非单独存在,需要通过特定的制备方法才能得到。
特戊酸氯甲酯是一种重要的医药中间体,常用于生产第二代口服头孢类抗生素,如头孢他内酯和头孢特质。此外,它也是中药原材料的一部分。然而,特戊酸氯甲酯本身并不存在,需要通过一系列的制备步骤来合成。
具体的制备方法如下:首先,在室温条件下,将三甲基乙酸(5.68mmol)溶解在二氯甲烷(15mL)和水(17mL)的混合溶液中。然后,在剧烈搅拌的条件下,缓慢加入碳酸氢钠(28.45mmol)和四丁基硫酸氢铵(0.56mmol)。接下来,在0℃下滴加氯甲基氯磺酸酯(8.5mmol),并在室温下搅拌反应24小时。反应完成后,向反应液中加入100mL乙酸乙酯,然后用饱和氯化铵水溶液洗涤,将水相用乙酸乙酯萃取,最后合并有机相。此外,还需要用蒸馏水和饱和氯化钠溶液依次洗涤有机相,然后使用无水硫酸钠干燥,并进行硅胶柱层析纯化,最终得到特戊酸氯甲酯。
以上就是特戊酸氯甲酯的制备方法。希望这些信息能对您有所帮助。
特戊酸氯甲酯是一种有机中间体,可通过一步制备得到。有文献报道其可用于制备替比培南酯和阿德福韦酯等原料药。
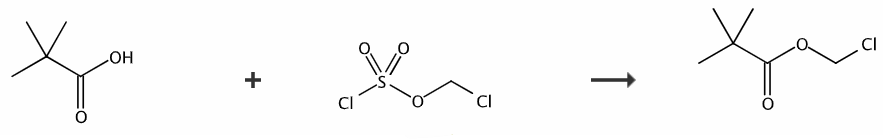
在室温条件下将三甲基乙酸溶解在混合溶液中,剧烈搅拌的条件下缓慢加入碳酸氢钠及四丁基硫酸氢铵,然后在0℃下滴加氯甲基氯磺酸酯,室温搅拌24h。反应完成后,进行后处理步骤,得到特戊酸氯甲酯。
CN201310335102.4报道了一种适合工业化制备替比培南酯的方法。该方法操作简便,避开了繁琐操作,后处理溶剂的使用量少,节约资源,降低环境污染,且特戊酸氯甲酯性质稳定,常温反应要求低,产品收率高,纯度达到药用要求。
CN201010124124.2提供了一种阿德福韦酯原料药的工业化生产合成工艺。该工艺简单、易操作、安全,环保,成本低,产品纯度高,可工业化生产。
CN201611144291.7公开了一种具有抗菌防辐射功能的胶原蛋白羊绒复合面料。该面料具有更好的抗菌防辐射性能和染色性能。
[1] CN201611144291.7一种具有抗菌防辐射功能的胶原蛋白羊绒复合面料及其制作方法
[2] CN201310335102.4一种工业化制备替比培南酯的方法
[3] CN201010124124.2一种阿德福韦酯原料药的合成工艺
[4] [中国发明] CN201810959516.7 取代吡唑类化合物、其制备方法、药物组合物及用途
特戊酸氯甲酯是一种常用于医药中间体的化学品,但本身具有一定的危险性,属于易燃物。在操作和储存时,必须远离火源。
在灭火过程中,必须佩戴自给式呼吸设备和全身防护服。首先,使用喷水迅速冷却暴露在空气中的火源容器,以防止可能的爆炸。火源的热量可能与酸或水分反应,产生爆炸性氢气,引发剧烈反应。对于冷却容器,应选择喷水方式。对于其他区域,应选择泡沫、二氧化碳或干粉灭火剂,但切勿使用直流水。
同时,应考虑意外泄漏的处理方式。对于小范围泄漏,建议使用干沙或泥土等惰性材料吸收泄漏物,并放入化学废物容器中。务必清除所有火源,并提供良好通风条件。
以上是关于特戊酸氯甲酯消防措施的介绍。正确处理燃烧或爆炸情况,可以避免更多的危害。
特戊酸碘甲酯,又称叔戊酸碘甲酯、新戊酸碘甲酯、2,2-二甲基丙酸碘代甲酯,是一种重要的医药中间体,可用于合成头孢卡品酯和头孢妥仑匹酯等产品。

目前,制备特戊酸碘甲酯的常用方法是在丙酮中使用碘化钠进行直接取代反应。虽然文献报道了高纯度和高收率的方法,但实际上很少能够达到这种水平。
本发明的创新点
本发明提供了一种收率大于90%的特戊酸碘甲酯制备方法,解决了现有技术中纯度低和收率低的问题,为后续产品的品质和成本提供了保证。
本发明采用以下技术方案:以特戊酸氯甲酯为原料,在乙酸乙酯溶剂中与碘化钠发生取代反应,反应回流时间为6小时。
此外,反应中还需加入氯化钙以控制过程水分。
具体操作步骤为:取特戊酸氯甲酯,加入乙酸乙酯作为溶剂,然后加入碘化钠和氯化钙,升温至78°C回流6小时,降温至0°C,用5%硫代硫酸钠洗涤至无色,再用无水硫酸镁干燥,最后减压浓缩即可得到特戊酸碘甲酯。
特戊酸氯甲酯、碘化钠和氯化钙的摩尔比为1:1.2~1.5:0.5~1.2。
与其他合成方法相比,本发明提供的特戊酸碘甲酯合成方法具有较高的摩尔收率。此外,该方法所得产品的纯度(GC)可达到98%,因此在成本和纯度方面具有显著优势。
具体实施方式
以下以实施例进行详细描述,但本发明的保护范围不限于以下实施例。
实施例1:在三口瓶中加入特戊酸氯甲酯10g、乙酸乙酯30mL、碘化钠11.6g和氯化钙3.6g,升温至78°C回流6小时,降温至0°C,然后用5%硫代硫酸钠洗涤至无色,再用无水硫酸镁干燥,最后减压浓缩。
反应摩尔收率为94%,GC:98%。
实施例2:在三口瓶中加入特戊酸氯甲酯10g、乙酸乙酯30mL、碘化钠13.4g和氯化钙4.6g,升温至78°C回流6小时,降温至0°C,然后用5%硫代硫酸钠洗涤至无色,再用无水硫酸镁干燥,最后减压浓缩。得到黄色液体即特戊酸碘甲酯化合物。
反应摩尔收率为90%,GC:97.5%。
特戊酰氯是一种化学品,具有遇水分解的特性,并且存在一定的风险性。因此,在操作过程中需要掌握正确的方式,并了解该物质可能对环境造成的危害,以避免对环境产生不良影响。
特戊酰氯在医药和农药领域具有广泛的应用前景。它可以作为中药的中间体,用于生产抗生素类药品。此外,特戊酰氯还可以应用于有机合成工业中。它是生产氨苄三水酸和羟氨苄三水酸等半合成抗生素的重要原材料,也是农药烯唑醇、烯效唑和多效唑等的基本原料。
特戊酰氯还可用作合成聚合引发剂过氧新戊酸叔丁基酯的原料,以及特戊酸氯甲酯的有机合成中的组成部分。随着国内对半合成抗生素品种的不断开发和增加,特戊酰氯的需求量也在不断增大,因此其应用前景非常可观。
通过以上介绍,我们可以了解到特戊酰氯具有非常广阔的应用前景,并且需求量也在逐渐增加。
新戊醇,即2,2-二甲基丙醇,是一种有机化合物,属于醇类。它是树脂状的结晶性固体,具有强刺激性,因此在使用时需要特别小心。

新戊醇作为广泛应用于农药、医药或电化学制品生产的重要有机化合物,有多种制备方法。以下是三种常见的方法:
1. 特戊酸及其衍生物的还原:特戊酸、特戊酸甲酯或乙酯、特戊酰氯和特戊酰胺在无水溶剂中用氢化锂铝(LiAlH4)还原。然而,这种方法的反应条件苛刻,设备要求较高且反应时间过长,且副反应产物较多。
2. 特戍醒还原:使用特戊醛还原制备新戊醇,但使用的催化剂Pt02昂贵且原料不易制备,导致整体成本过高。
3. 格氏试剂与甲醛进行加成反应:使用叔丁基氯化镁和低聚甲醛在四氢呋喃溶剂中进行加成反应来制备新戊醇。然而,该方法操作复杂,产品纯度较低,无法满足高端产品的需求。
为了克服上述方法的不足,我们提出了一种新的合成方法。该方法使用金属钠与相应的酯反应,反应过程中没有副反应生成,可以得到高含量的新戊醇。该方法不需要特殊催化剂或设备,成本较低,适合工业化生产。具体的实施方法是将特戊酸酯与溶剂A及所用醇混合,滴加到盛有金属钠的反应容器中,在50-90°C进行反应,直到酯完全反应。然后蒸去过量的醇,用水酸化后经精馏获得高纯度的新戊醇产品。
在合成方法中,金属钠用量为酯用量的5-8倍(摩尔),醇用量为酯用量的20-50倍(摩尔),溶剂用量为酯用量的1-5倍(摩尔),反应温度为50-90°C。
反应方程式:

对苯二甲醛是一种重要的有机合成和精细化工原料,广泛应用于染料、荧光增白剂、医药和香料等工业领域。它具有两个活泼醛基,可以自聚或与其他单体共聚形成各种高分子化合物,因此也是合成高分子材料的重要单体。

氯化水解法是对苯二甲醛最传统的合成工艺路线。首先对二甲苯进行氯取代,然后通过水解或氧化水解得到对苯二甲醛。氯化反应转化率高,水解过程受催化剂影响,产率有所差异。在氯化水解后,需要经过碱中和、过滤干燥才能得到对苯二甲醛粗品。除了目标产物对苯二甲醛,还会生成副产物对醛基苯甲酸等。
为了避免直接使用对环境不友好的氯气,可以先采用N-氟代双苯磺酰胺和氯化钠与对二甲苯反应,合成对二氯苯,然后通过硫酸和二氧化锰催化作用得到对苯二甲醛粗品。最后通过氢氧化钠中和氧化母液,乙醇水混合液中重结晶,得到对苯二甲醛成品,重量收率为77%。
对苯二甲酸(酯)加氢是另一种合成对苯二甲醛的方法。催化剂的选择非常重要,过高的加氢活性会导致大量芳香醇生成。有效的催化剂体系包括钯的络合物、铬系金属氧化物和硼的氟化物。采用钯的三苯基膦络合物或钯的三苯基膦与钯的乙酸酐盐复合体系作为催化剂,在适当的温度和压力下,可以催化苯甲酸形成苯甲醛,也可以催化芳烃二酸加氢生成醛。但该催化剂需要在严格无氧环境和高压釜中反应,并且产物需要从大量的特戊酸体系中分离,能耗较高。
在特定条件下,采用Cr-ZrO2催化剂可以实现对苯二甲酸和对醛基苯甲酸甲酯的高选择性加氢转化。另一种Zn-ZrO2催化剂对苯二甲醛的选择性更高,但转化率较低。
酰氯化合物加氢脱氯生成醛是一种重要的反应,通常采用Pd/BaSO4作为催化剂。近年来,研究发现在多甲基羟基硅烷、氟化钾、三(2-呋喃基)膦等共同作用下,Pd的金属络合物能够高效催化苯甲酰氯生成苯甲醛,该催化体系也适用于其他芳烃酰氯化合物。
采用Pd负载量为3 wt%的Pd/SiO2催化剂,在适当的温度和反应时间下,可以高效转化对苯二甲酰氯为对苯二甲醛和对甲基苯甲醛等醛类产品。
喹啉衍生物是一类具有多种生物活性和药理活性的化合物,包括杀菌、抗菌、抗高血压、抗抑郁、抗过敏、抗疟疾、抗肿瘤和抗癌等作用。本文介绍了一种制备6-乙酰基喹啉的方法。

首先,在杨氏反应管中准确称取0.005mmol(1.1mg)过渡金属,并加入10mL已放入磁力搅拌子的杨氏反应管内。然后,进行氧气置换,使反应在氧气条件下进行。接下来,使用注射器向杨氏反应管中准确加入0.04mmol辅助催化剂I、0.08mmol辅助催化剂II、0.2mmol 4-乙酰基苯胺和1ml 1,3-丙二醇。将杨氏反应管置于磁力搅拌器上,在150℃下搅拌12小时。反应结束后,调节反应液的pH为中性,并对反应溶液进行后处理,得到纯品6-乙酰基喹啉,产率为48%。
该方法中使用的金属催化剂可以是氯化钯、乙酸钯、四三苯基膦钯、三(二亚苄基丙酮)二钯、氯化烯丙基钯(II)二聚物或三氟乙酸钯等。金属催化剂的用量为2-5%mol。辅助催化剂I可以是吡啶、2-吡啶甲酸、2-二联吡啶、3-吡啶甲酸乙酯、2-甲氧基吡啶、4-(N,N-二甲基)吡啶、喹啉、2-(N-甲基)吡啶胺、4-甲基-2-甲氧基吡啶、2,3,4-三甲基吡啶、2-甲基吡啶、2-甲基喹啉、1,10-邻菲咯啉、2-(N,N-二甲基)吡啶、2,4-二甲基吡啶、3-吡啶甲基酮、2,5-二甲基吡啶、2,6-二甲氧基吡啶、2,4-二甲氧基吡啶、2-吡啶甲酸乙酯、2,6-二异丙基吡啶或2-羟基吡啶等。辅助催化剂I的用量为5-10%mol。辅助催化剂II可以是对甲苯磺酸、特戊酸、三氟乙酸、乙酸、甲磺酸、三氟甲磺酸或醋酸酐等。辅助催化剂II的用量为10-20%mol。

步骤1:制备N-甲氧基-N-甲基喹啉-6-甲酰胺(B-2)
将喹啉-6-羧酸B-1(5g,29mmol)溶于无水DMF(60mL)中,氮气保护下加入2-(7-偶氮苯丙三氮唑-1-基)-N,N,N’,N’-四甲基脲六氟磷酸酯(12.9g,34mmol)和盐酸二甲基羟胺(3.3g,34mmol),室温搅拌过夜。加入二氯甲烷(80mL)和水(80mL),分出有机相,用二氯甲烷(50mL×3)萃取,合并有机相,用饱和氯化钠(50mL×2)洗,干燥。蒸干得到4.6g无色油状液体化合物B-2,收率为74.5%。
步骤2:制备6-乙酰基喹啉(B-3)
将N-甲氧基-N-甲基喹啉-6-甲酰胺B-2(6.1g,28mmol)溶于无水THF(60mL)中,冰浴冷却到0℃,氮气保护下加入3M甲基溴化镁(10mL)溶液。撤去冰浴,室温搅拌过夜。反应液中缓慢加入饱和氯化铵水溶液(12mL),加入乙酸乙酯(50mL)和水(30mL),分出有机相。水相用乙酸乙酯(50mL×3)萃取。合并有机相,用饱和氯化钠溶液(50mL×2)洗,干燥。蒸干得到4.4g淡黄色固体化合物B-3,收率为91.8%。
[1] [中国发明,中国发明授权] CN201610578354.3 一种喹啉衍生物的制备方法
[2] [中国发明,中国发明授权] CN201410152877.2 喹啉类化合物、其制备方法、中间体、药物组合物和应用
引言:
研究如何利用N-[(S)-乙氧羰基-1-丁基]-(S)-丙氨酸合成培哚普利及其相关化合物,对于提高此类药物的合成效率和质量具有重要意义。优化合成方法不仅能推动新药研发,还能促进其在临床应用中的普及。
简介:
N-[(S)-乙氧羰基-1-丁基]-(S)-丙氨酸,英文名称:N-[(S)-Ethoxycarbonyl-1-Butyl]-(S)-Alanine,CAS:82834-12-6,分子式:C10H19NO4,外观与性状:白色或淡黄色结晶粉末,密度:1.079 g/cm3,沸点:324.081℃ at 760 mmHg,熔点:145-150℃。N-[(S)-乙氧羰基-1-丁基]-(S)-丙氨酸是培哚普利的重要中间体。以正戊酸为原料,经卤化、缩合、还原等5步反应可得到N-[(S)-1-(乙氧羰基正丁基)]-S-丙氨酸。
应用举例:合成培哚普利
培哚普利(Perindopril),作为无巯基的ACE抑制剂,主要用于治疗原发性高血压与心力衰竭,于1989年在法国首次上市.它不仅具有其他ACE抑制剂的优点,而且作用时间更长,副作用更小,耐受性更好,具有良好的市场前景。
(1)报道一
杨新华等人以(2S,3αS,7αS)-八氢吲哚-2-羧酸和N-[(1S)-(1一乙氧羰基)-丁基]-(S)-丙氨酸为原料,经酯化、缩合、氢化3步反应合成培哚普利叔丁胺盐,总收率为46%。
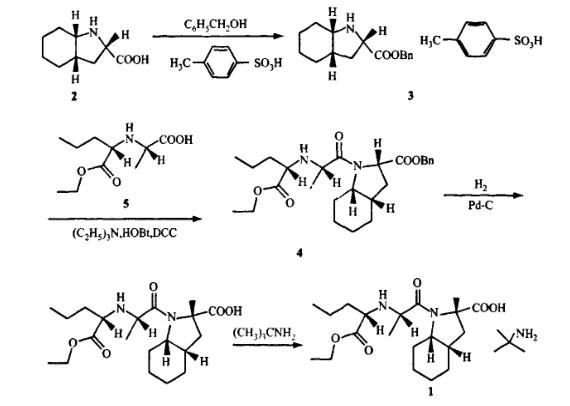
(2)报道二
高毅平等人以(2S,3aS,7aS)-八氢吲哚-2-羧酸苄酯对甲苯磺酸盐与N-([S]-1-乙氧羰基丁基)-[S]-丙氨酸经缩合、催化加氢、成盐后,即得产品培哚普利叔丁胺盐。该合成周期缩短,总收率提高16%,纯度由94.26%提高到99.51%。
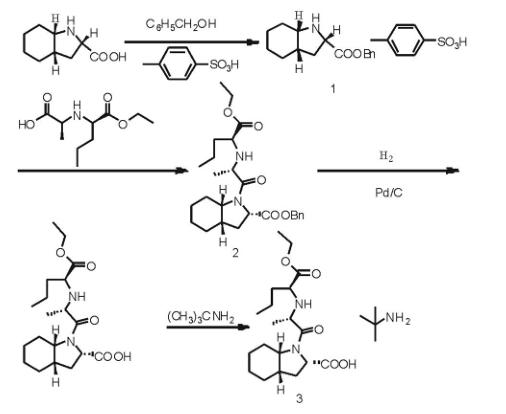
(3)报道三
张红娟等人以N-[(S)-1-(乙氧羰基)丁基]-(S)-丙氨酸和(2s,3aS,7aS)-八氢吲哚-2-羧酸为原料,用氯甲酸甲酯将N-[(S)-1-(乙氧羰基)丁基]-(S)-丙氨酸的氨基保护,经亚硫酰氯活化,与(2s,3aS,7aS)-八氢吲哚-2-羧酸反应,再与叔丁胺成盐,最终制得培哚普利叔丁胺盐。
(4)报道四
在沈阳药科大学所报道的文章中,以(2,3,7)-八氢-吲哚-2-羧酸和N-[(S)-1-(乙氧羰基正丁基)]-S-丙氨酸为中间体的合成路线,对培哚普利进行合成。其中,以(S)-二氢吲哚-2-羧酸为原料,经还原反应,合成(2,3,7)-八氢-吲哚-2-羧酸。以正戊酸为原料,经卤化、缩合、还原等5步反应合成N-[(S)-1-(乙氧羰基正丁基)]-S-丙氨酸。两个重要中间体缩合、脱保护和成盐等反应得到培哚普利特丁胺盐。
参考:
[1] 杨新华. 培哚普利叔丁胺盐的合成[J]. 化学世界,2010,51(9):552-554. DOI:10.3969/j.issn.0367-6358.2010.09.012.
[2] 高毅平,朱雍,唐伟方,等. 培哚普利的合成工艺研究[J]. 海峡药学,2012,24(10):255-257. DOI:10.3969/j.issn.1006-3765.2012.10.140.
[3] 培哚普利(Perindopril)的合成工艺[Z]. 沈阳药科大学. 2005.
[4] 张红娟. 培哚普利叔丁胺盐的制备及氧化氯化法提高甲苯氯化产品的对位选择性[D]. 江苏:南京大学,2012.
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手




























