
肉桂醛二乙缩醛在碱性介质中比较稳定,可作为花香型皂用,洗涤剂香精的头香剂和修饰剂,亦可用于柑橘型食用香精中。
一种肉桂醛二乙缩醛的合成方法,反应式如下:
将三苯基磷10g、甲苯111g、苯甲醛0.4mol和2-溴-1,1-二乙氧基乙烷0.35mol加入到平底烧瓶中,充分混合后倒入装有负载KOH的大孔树脂载体的管式反应器中,放入微波炉中,装上温度计和冷凝器;在搅拌常压状态下进行微波照射,功率550W,升温至88℃,并保温回流1.5h;反应完毕,反应结束后冷却到室温,得到反应液,然后常压蒸馏出溶剂甲苯,过滤出催化剂三苯基膦,然后反应液进一步经过刮板式分子蒸馏装置蒸馏出产物肉桂醛二乙缩醛。
本发明采用原料价格便宜易得,催化剂为固体催化剂易于除去,反应条件温和简单,副反应少,后处理容易,产品收率和纯度高,成本大大降低,可连续化生产。
CN102701924B
4-硝基肉桂醛是一种有机中间体,可以通过一步反应从对硝基苯甲醛和乙醛制备而成。

将5.0克(66mmol)对硝基苯甲醛和10.0毫升乙醛加入三颈瓶中,然后在滴液漏斗中加入10毫升质量分数为30%的氢氧化钾-甲醇溶液。在磁力搅拌下,用冰盐浴冷却反应瓶,当温度降到0-5℃时,缓慢滴加5毫升氢氧化钾-甲醇溶液,反应40分钟后再滴加5毫升氢氧化钾-甲醇溶液。在10℃左右继续反应40分钟,冷却至5℃以下时,反应混合物固化。然后加入16毫升乙酸酐,在水浴上加热30分钟,倒入120毫升热水中,加入16毫升浓盐酸,再在水浴上加热20分钟,静置24小时。最后,将析出的晶体抽滤、洗涤,并在质量分数30%的乙酸水溶液中进行重结晶,得到1.6克黄色晶体,即为4-硝基肉桂醛,产率为45.3%。
在室温下,氩气气氛下,将丙烯醛二乙缩醛(3当量)、K2CO3(1.5当量)和Pd(OAc)2(3 mol%)加入到搅拌的p-取代碘/溴苯(0.100克)在密封的干燥DMF(2毫升)溶液中,将反应混合物在90°C下照射10分钟(1个循环)。冷却反应混合物,小心地将其倒入2N HCl水溶液中,并在磁力搅拌下静置10分钟。使用乙醚萃取水层,然后用硫酸钠干燥有机层。在减压下浓缩有机层,并使用适当的洗脱液通过柱色谱法纯化残余物,最终得到4-硝基肉桂醛。
[1] [中国发明,中国发明授权] CN201610146654.4 一种富勒烯吡咯烷衍生物及其制备方法
[2] From Tetrahedron Letters, 48(28), 4801-4803; 2007
托特罗定是一种有效的药物,被广泛应用于现已被广泛用于尿急、尿失禁等症状,在本文中,我们将探讨如何合成这种重要药物。
简介:托特罗定(tolterodine?PNU-200583)是Phar macia& Upjohn公司研制开发的新型M受体拮抗剂。1998年首先在瑞士上市,现已被广泛用于治疗膀胱逼尿肌过度兴奋引起的尿频、尿急、尿失禁症状,其作用特点是比现在临床上使用的奥西布宁副作用的发生率低得多。该药化学名为(+)-N,N-二异丙基-3-(2-羟基-5-甲基苯基)-3-苯基丙胺-(+)-酒石酸盐,商品名为Detrol。
合成:
1. 有研究以肉桂酸和对甲酚为起始原料经环合、甲基化、氯化、酰胺化、还原、脱甲基、拆分7步反应制备托特罗定酒石酸盐,总收率为14.4%。合成路线如图所示。

化合物4的制备中,最佳反应条件:不用四氢萘作溶剂,将肉桂酸与对甲酚直接混合,肉桂酸-对甲酚-浓硫酸的摩尔质量比为1∶1.05∶0.4,反应6 h,收率为91%。在化合物8的制备中,故选用还原剂如NaBH4-浓硫酸、NaBH4-POCl3、Na-正丁醇、KBH4-ZnCl2-甲苯体系进行试验,均没有得到较为满意的结果。又对LiAlH4的用量进行考察,当化合物7与LiAlH4的配比为1∶2.4,回流反应80 h,TLC检测转化得的很少。当其配比提高到1∶3.2时,仅12 h,收率可达到 84.5%。由于乙醚沸点较低,改为THF作溶剂, 反应10 h,收率为81%。
2. 托特罗定及其对映体的合成
1. 专利CN 110229072 A公开了一种托特罗定及其对映体的合成方法,属于化学合成领域。该发明以肉桂醛为原料,在铑催化剂的作用下,与(2-羟基-5-甲基苯基)硼酸发生不对称芳基化反应,得到的半缩醛中间体无需纯化,直接进行还原胺化反应即可得到高光学纯的托特罗定。以2-羟基-5-甲基肉桂醛和苯硼酸为起始原料,使用相同的合成方法和操作步骤,即可制备高光学纯的托特罗定对映体。该发明报道的方法具有路线简短、总收率高和立体选择性好等特点,合成的两种构型产物的ee值均大于99%。

具体步骤:(1)依次将肉桂醛、(2-羟基-5-甲基苯基)硼酸、铑催化剂和碱加入Schlenk管中,在氮气保护下加入第一种溶剂,20~100℃下搅拌反应5~20h;随后将反应溶液倒入柱层析中,并用乙酸乙酯冲洗;将溶液减压蒸除,并加入第二种反应溶剂,搅拌下加入二异丙基乙胺、氰基硼氢化钠和四异丙氧基钛,加毕,20~80℃继续反应4~15h;去除反应液,固体残渣经柱层析纯化,得到化合物I;
(2)以2-羟基-5-甲基肉桂醛和苯硼酸、铑催化剂和碱加入Schlenk管中,在氮气保护下加入第一种溶剂,20~100℃下搅拌反应5~20h;随后将反应溶液倒入硅胶柱中,并用乙酸乙酯冲洗;将溶液减压蒸除,并加入第二种反应溶剂,搅拌下加入二异丙基乙胺、氰基硼氢化钠和四异丙氧基钛,加毕,20~80℃继续反应4~15h;去除反应液,固体残渣经柱层析纯化,得到化合物II。
2. 托特罗定及其对映体的早期合成依赖于手性拆分的方法,如专利WO2007147547A1公开的合成方法如下:

但该方法以肉桂基氯为原料,经三步反应得到消旋体,最后经L-酒石酸拆分得到单一对映异构体。手性拆分需要大量的拆分试剂,原子经济性差,且操作繁琐。
3. 为实现托特罗定及其对映体的不对称合成,近年来,研究人员开发了一系列不对称催化合成的方法,如J.Org.Chem.,2007,72:6056-6059报道了不对称合成化合物II的方法,如下所示:

该方法以2-溴-4-甲基苯酚为原料,经四步反应得到化合物II。该合成路线的总收率为47.2%,但终产物的ee(80%)值欠佳。
参考文献:
[1]周辛波,高文芳,郑红等.M受体拮抗剂托特罗定的合成研究[J].中国药物化学杂志,2002(02):47-49.
[2]中国药科大学. 一种托特罗定及其对映体的合成方法:CN201910553495.3[P]. 2019-09-13.
本文介绍了如何使用5-氟-1-茚酮来合成茚虫威衍生物,旨在为相关研究人员提供参考依据。
简述:5-氟-1-茚酮,英文名称:5-Fluoro-1-indanone,CAS:700-84-5,分子式:C9H7FO,外观与性状:灰白色晶体粉末,折射率:1.459。
应用:合成茚虫威衍生物。
茚虫威(Indoxacarb)是由美国杜邦公司开发的一类对鳞翅目以及部分同翅目和鞘翅目害虫具有优良杀虫活性的噁二嗪类杀虫剂。
当茚酮环上的5位取代基为吸电子基团时,对昆虫钠离子通道的抑制率和杀虫活性表现出良好效果。基于这一发现,张健强选择了5-氟-1-茚酮、5-氯-1-茚酮和5-溴-1-茚酮作为起始原料,合成出具有卓越杀虫活性的DCJW类似物G1-G4。随后,通过将茚酮侧链上的甲氧甲酰基还原为羟甲基,得到醇类化合物H1-H4。这些化合物与拟除虫菊酰氯、肉桂酰氯、噻吩乙酰氯和氯乙酰氯等酰氯在氩气保护、无水THF中经过DMAP催化反应,合成得到多种化合物。具体步骤如下:
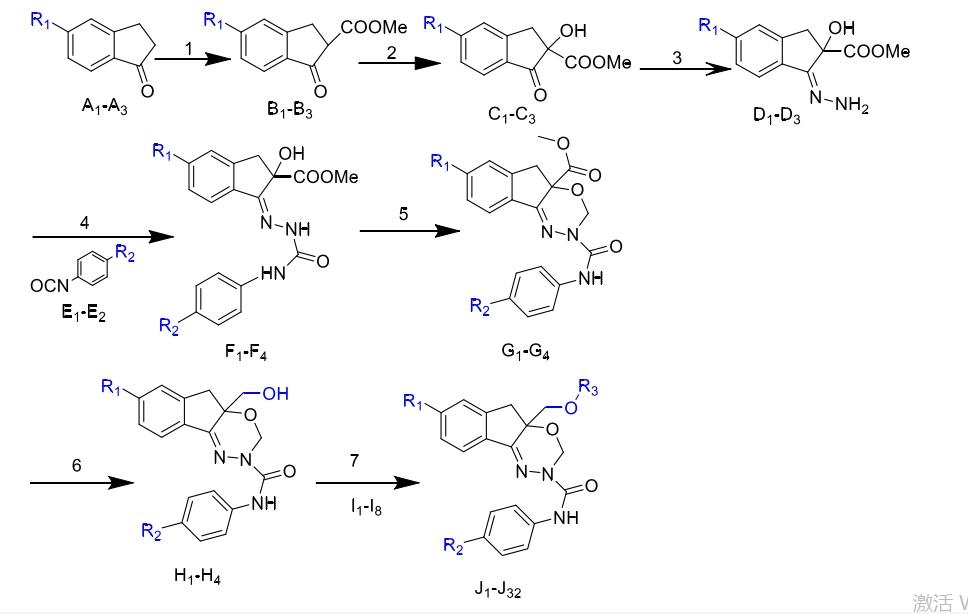
(1)在冰浴,氩气保护条件下,在250 mL 的三口瓶中加入KTB(4.2 g,37.5 mmol,1.25 eq)和NaH(3.03 g,120.0 mmol,4eq), 混合均匀。将干燥DMC(30 mL)加到混合物中,匀速搅拌5 min后至混合液变为浅灰色,再用注射器将化合物A(30 mmol,1.0eq)的干燥DMC(70.0 mL)溶液匀速缓慢滴加到以上混合液中(反应体系由浅灰色变为红色,随着反应进行,析出大量固体,由红色逐步转化成墨绿色)升温至室温反应进行2 h。用冰HCl猝灭,并调pH至4-5,EtAc(3 × 50mL)萃取,分液,有机层用饱和NaCl溶液洗涤至中性,无水 MgSO4 干燥,过滤,脱溶,得到棕色固体化合物B1-B3的粗产品,粗产品无需处理直接进行 下一步反应。
(2)在室温氩气保护下,在250 mL的圆底烧瓶中将含有化合物A(30.0 mmol,1eq)的甲苯(60.0 mL)溶液用注射器滴加至辛可宁(886 mg,30 mmol,0.1eq)中,充分搅拌5 min,再将65%的t-BuOOH溶液(5.1 mL,33 mmol,1.1eq)用注射器逐滴加入以上混合液中,反应3 h左右有固体析出,室温反应 24 h。TLC跟踪,至化合物A基本消失。直接抽滤,用少量甲醇的50%水溶液冲洗,干燥得白色粗品,滤液用EtAc(3 × 50 mL)萃取,有机层用无水 MgSO4干燥、浓缩,进行硅胶柱层析(Petroleumether/Ethylacetate = 4:1–1:1,v/v),得到白色粉状固体化合物C1-C3。
(3)在冰浴条件下,将95%冰乙酸(5.2 mL,90 mmol,3eq)滴加至含有85%水合联氨(5.1 mL,90 mmol,3eq)的MeOH(30 mL)溶液中,充分搅拌5min,再将含化合物C(30 mmol,1eq)的MeOH(120 mL)溶液 滴加至上述混合溶液中,升温到90℃回流反应进行3.5 h。TLC跟踪,反应结束后将 反应液浓缩去除绝大部分MeOH后,加入饱和NaCl溶液,再用(3×100 mL)EtAc 萃取,无水 MgSO4干燥、过滤、脱溶得到油状化合物D1-D3粗产物,直接进行下一 步反应。
(4)在氩气保护,室温条件下,将化合物E(6 mmol,1.2eq)滴加至化合物D(5 mmol,1.0eq)的干燥THF(20 mL)溶液中。 1 min后有白色沉淀出室温反应2 h后,将获得溶液直接抽滤,用 (Petroleumether/Ethylacetate = 6:1,v/v)的混合液(3×15 mL)洗涤浓缩物,滤液浓 缩,进行硅胶柱层析(Petroleumether/Ethylacetate = 2:1,v/v),将滤饼与过柱纯化得到 的化合物合并,得到白色固体化合物F1-F6。
(5)在室温氩气保护下,在250 mL三口瓶中加入P2O 5(550 mg,3.75 mmol,2.5eq)和硅藻土(550 mg),将干燥的1,2-DCE(30 mL)和甲缩醛(12.5 mL)分别滴加至上述混合物中,室温搅拌30 min,待P2O5和硅藻土充 分分散,再将含有化合物F(1.54 mmol,1eq)的1,2-DCE(20 mL)溶液用注射器缓慢匀速滴加到以上反应液中,升温至57℃反应每进行2 h,分别补加硅藻土(550 mg)、 P2O5(110 mg,0.75 mmol,0.5eq)、甲缩醛(12.5 mL)直到原料反应消耗完为止。TLC 跟踪反应。反应结束后,用H2O猝灭,NaHCO3中和,有机相用无水MgSO4干燥、过滤,滤液为棕黄色油状物,加入石油醚(10 mL)重结晶,4℃下静止放置10 h,析出固体过滤得到滤饼烘干,滤液浓缩进行硅胶柱层析(Petroleumether/Ethylacetate =6:1–4:1,v/v),所得化合物合并,得到化合物G1-G6。
(6)在冰浴氩气保护下,在100 mL的圆底烧瓶中将化合物 H1-H6(2 mmol,1eq)的干燥THF(10 mL)溶液滴加至 LiAlH4(38 mg,1 mmol,0.5eq) 的干燥THF(5 mL)悬浮液中,升温至室温反应,每过2 h分别补加LiAlH4(38 mg, 1 mmol,0.5eq)并对反应进行TLC跟踪,直至原料完全耗尽。所得混合液直接加入硅 胶浓缩干燥,进行硅胶柱层析纯化(Petroleum ether/Ethyl acetate = 2:1–1:2,v/v),得到化合物H1-H6。
(7)反应操作步骤:在冰浴氩气保护下,将干燥的DIPEA(0.42 mL,3 mmol,3eq)滴加至化合物H(1 mmol,1eq)和DMAP的混合干燥THF(5 mL)溶液中,再将酰氯I1-I8(3 mmol,3eq)滴加至其中。升温至室温反应2 h之后,有沉淀析出,所得混合液用冰Na OH(3 × 30 mL)溶液洗涤三次,用乙酸乙酯(3 × 15 mL)萃取,无水硫酸镁干燥,收集洗涤液浓缩进行硅胶(300-400目)柱层析(Petroleum ether/Ethyl acetate = 8:1,v/v),与二氯菊酸偶联的化合物J1-J4、功夫菊酸J5-J8和氰戊菊酸J29-J32 偶联的化合物由于结构特殊,在TLC上显示距离相近的两个斑点。通过柱层析可以分离得到两个低极性的化合物H-Rf和高极性的化合物L-Rf。
参考文献:
[1]张健强.茚虫威衍生物的设计合成、生物活性及分子对接研究[D].华南农业大学,2018.
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手






















