针对4-溴苯甲酸的合成,目前存在着多种方法,本文将介绍其中两种常见且有效的合成方法,以期为相关领域的研究者提供参考。
简介:4-溴苯甲酸(4-Bromobenzoic acid) 又称作对溴苯甲酸,为白色或浅粉红色结晶。4-溴苯甲酸是重要的精细化学品,可用作香料原料、4-溴苯甲酸酯的中间体、测量锶、有机微量分析测碳、氢和溴的标准品。目前4-溴苯甲酸的生产方法主要有两种,一种是由高酸钾氧化对溴甲苯制得对溴苯甲酸,成本太高,且收率只有80%:另一种方法是由溴苯与次氯酸钠反应生成4-溴苯乙酮,然后再将4-溴苯乙酮与次氯酸钠反应生成4-溴苯甲酸,该方法反应步骤多,污染严重。针对上述现有技术存在的问题,待研究开发出一种污染小、成本低、收率高的制备4-溴苯甲酸的新工艺。
合成:
1.1 方法一:用高锰酸钾氧化法制备苯甲酸类化合物,在以往的文献中大多采用了中性条件来进行实验,但是在实验中发现,中性条件不能达到预期的效果。有研究考察了碱性、酸性条件下,以对溴甲苯为原料,经相转移催化,由高锰酸钾氧化合成4-溴苯甲酸的反应。
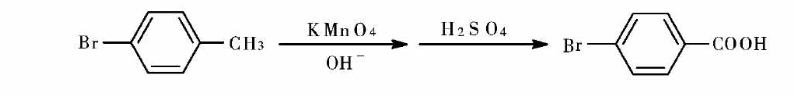
1.2 依次向三颈瓶中加入对溴甲苯、催化剂TEBA 、水,打开回流冷凝装置和搅拌器.水浴加热,待水浴温度升至一定温度,向三颈瓶中分批加入高锰酸钾,加完后,控温反应时间2.5 h 左右,反应完毕,趁热抽滤,除去反应中生成的沉淀.滤饼再用沸水连续洗涤2 次,把洗涤液与滤液合并.如液体的颜色为紫色,则加入乙醇,加热使高锰酸钾分解,再过滤,洗涤。
将合并的滤液浓缩,冷却后加入50 %的硫酸进行酸化,出现白色沉淀,放置冷却后,抽滤,滤饼用冷的蒸馏水洗涤,放进红外恒温干燥箱进行干燥,称重。熔点:251 ~ 256 ℃。
1.3 通过一系列的实验发现,采用相转移催化剂、用高锰酸钾氧化对溴甲苯合成4-溴苯甲酸适宜的工艺条件为n(对溴甲苯):n (KMn04):n(Na2CO3)为1.0:2.5:1.0,TEBA用量为5%,反应温度95C左右反应时间2.5h,产率为82.4%。使用该方法制备对溴甲苯酸,反应条件温和,时间短,操作简单,生产成本低.对设备的腐蚀小,对环境的污染少。
2.方法二:另一种4-溴苯甲酸的制备方法,是以对溴甲苯作为起始原料,冰醋酸为溶剂,氧气为氧化剂,利用液相氧化法在催化剂作用下催化氧化对溴甲苯,控制反应温度为75~85℃,当反应体系中的对溴甲苯的含量为初始含量的0.5wt%以下时结束反应,然后冷却过滤得4- 溴苯甲酸粗品和滤液,再将所述4-溴苯甲酸粗品进一步纯化制得成品。
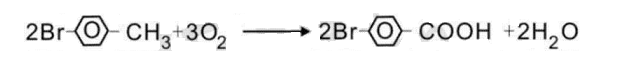
采用该发明方法制得的4-溴苯甲酸收率高,可达98%以上,同时成品纯度也高,可达99%以上,克服了传统制备方法制备该产物纯度低,收率低的缺陷,且该发明方法采用的原料简单易得,成本低,应用前景广阔。
参考文献:
[1]. CN201810447513.5 一种4-溴苯甲酸的制备方法
[2].刘荣,戚小露,支三军.相转移催化合成对溴苯甲酸的研究[J].淮阴师范学院学报(自然科学版),2011,10(04):316-318.DOI:10.16119/j.cnki.issn1671-6876.2011.04.003.
[3].吴育飞,胡瑞省.间接电合成对溴苯甲酸[J].河北化工,2005(01):19-20+25.
慢性阻塞性肺病(COPD)是一种常见的呼吸系统疾病,其患病率和病死率呈逐年上升的趋势,每年受其困扰的人多达6亿。到2020年COPD将成为全球第三大致死疾病。格隆溴铵作为一个新型长效抗胆碱能药物,能够产生更持久的支气管保护和支气管扩张作用,起效快,支气管扩张作用可持续24小时以上。格隆溴铵能够减少慢性阻塞性肺病患者的呼吸急促、病情恶化和急救药物的使用,有效改善肺功能,提高生活质量及运动耐受性。在中度至重度慢性阻塞性肺病患者中格隆溴铵显示有良好的安全性和耐受性。2-环戊基-2-羟基苯乙酸是合成格隆溴铵的重要中间体,它的合成方法是:苯甲酰甲酸或苯甲酰甲酸酯与卤代环戊烷的格式试剂反应。该反应副产物较多,得到的粗品纯度低。

文献Org.Process Res.Dev.2012,16,1754-1769对2-环戊基-2-羟基苯乙酸的合成方法做了详细的阐述,产生了较多的杂质,鉴于该反应的极限性,急需开发一种高效的2-环戊基-2-羟基苯乙酸精制纯化方法,并能够应用于工业化生产。
先制备环戊基格氏试剂,然后在20L四口瓶中加入495g苯乙酮酸,2.5L四氢呋喃,在15℃~25℃滴加环戊基格氏试剂2.1eq,滴毕在反应完后用4N盐酸淬灭,之后用MTBE萃取,碱洗,食盐水洗,浓缩得到中间体2-环戊基-2-羟基苯乙酸油状物563g。
向由上述反应所得油状物中加入150g乙醇,纯化水300g,升温至55~65℃使其溶解后,缓慢降温至0~5℃,保温1h后,抽滤,得棕黄色固体:将湿品再用227g乙醇,纯化水300g,升温至70~80℃溶解后,在缓慢降温至0~5℃,保温1h后,抽滤,得淡黄色固体;将湿品用300g乙醇,纯化水160g,升温至75~85℃溶解后,在缓慢降温至0~5℃,保温1h后,抽滤,得类白色固体214g;将粗品用1177ml甲苯升温至60~80℃溶解后,缓慢降温至0~5℃,保温搅拌1h后抽滤,得类白色固体,将湿品再用1177ml甲苯升温至90~110℃溶解后,缓慢降温至5~15℃,保温1h后抽滤,烘干后,得类白色固体2-环戊基-2-羟基苯乙酸171.36g,收率为23.6%,HPLC纯度为98.5%.
[1]上海奥博生物医药技术有限公司,浙江华海药业股份有限公司. 一种格隆溴铵中间体2-环戊基-2-羟基苯乙酸的纯化方法:CN201710413117.6[P]. 2018-12-11.
利莫那班是一种新型减肥药,而2-溴-3'-氯苯丙酮是合成利莫那班的关键中间体。2-溴-3'-氯苯丙酮是一种有机化合物。下面是对其性质、用途、制法和安全信息的介绍。
2-溴-3'-氯苯丙酮的分子式为C9H8ClOBr,分子量为247.5。它是一种无色或微黄色结晶固体,熔点为76-77℃。可溶于有机溶剂如乙醇、乙醚和氯仿,略溶于水。基于其结构,我们设计了一种改进的合成工艺路线,该方法使用金属卤化物作为催化剂,对液溴和对氯苯丙酮进行反应合成2-溴-3'-氯苯丙酮。这种方法具有反应速度快、反应时间短、反应选择性高以及产品收率和纯度较高的优点。
2-溴-3'-氯苯丙酮是合成利莫那班的关键中间体。它也可以用作催化剂、氧化剂和还原剂的配体。
2-溴-3'-氯苯丙酮可以通过以下反应制得:将2-溴苯乙酮与氯乙酸反应,生成2-溴-3'-氯苯丙酮[1].

图1 2-溴-3'-氯苯丙酮的合成反应式
首先,在装有回流冷凝管和滴液漏斗的反应器中加入镁粉和四氢呋喃溶液,然后缓慢加入溴乙烷,控制反应液温度并加热回流,得到Grignard试剂。接下来,将间氯苯甲腈滴加到格氏试剂中,反应生成中间体。最后,用盐酸水解中间体,分离有机相,经蒸馏得到3-氯苯丙酮。在另一个反应瓶中,加入3-氯苯丙酮、液溴、溶剂和催化剂,装上回流冷凝管和温度计,控制温度并进行反应。反应结束后,蒸馏出溶剂,洗涤剩余溶液,再用精制溶剂溶解,降温结晶,过滤得到精制的2-溴-3'-氯苯丙酮产品,收率达到97.13%,熔点为76-77℃.
2-溴-3'-氯苯丙酮具有刺激性,应避免与皮肤和眼睛接触。在操作时,应在通风良好的地方进行,避免吸入其蒸气。存储时应密封保存于干燥、阴凉的地方,并远离火源和可能的氧化剂。使用前请参考相关安全数据表(MSDS)获取详细的安全信息.
[1]张竞, 陈声宗, 吴灿, & 石变芳. (2002). 3-氯-2′-溴苯丙酮的合成研究. 化学试剂(06), 41-42.
RS-扁桃酸,化学名α-羟基苯乙酸,又称苦杏仁酸,具有强大的抗菌作用,可用于治疗泌尿系统感染疾病。此外,扁桃酸及其衍生物在精细化工中间体、医药生产等领域也有广泛应用。S-扁桃酸(右旋扁桃酸)是一种具有光学活性的扁桃酸,除了具备扁桃酸的应用范围外,还主要用于不对称合成和光学拆分。

RS-扁桃酸及其衍生物是一种重要的β-内酰胺侧链修饰剂,在头孢孟多等抗生素的生产中得到广泛应用。此外,扁桃酸还可用于合成环扁桃酯(血管扩张剂)、扁桃酸乌洛托品(尿路消毒剂)、苯异妥因(抗抑郁剂)、扁桃酸苄酯(镇痉剂)等药物的原料。具有单一构型的扁桃酸及其衍生物是不对称合成中重要的手性中间体,被广泛应用于光学纯的氨基酸、血管紧张肽转化酶抑制剂、辅酶A的合成。例如,S-扁桃酸是合成用于治疗尿急、尿频和尿失禁药物S-奥昔布宁的前体材料。
一种制备S-扁桃酸的方法包括以下步骤:
1) 在甲苯中通入卤素蒸汽,无需其他溶剂,加热回流,然后对反应液进行分馏,得到卤化甲苯。
2) 将卤化甲苯与氰化盐反应,生成苯乙腈。
3) 经过溴化处理,得到α-溴-苯乙酮,经酸化后得到RS-扁桃酸粗品。
4) RS-扁桃酸粗品在酸性环境下与右旋拆分剂反应生成S-扁桃酸盐酸盐,经过后处理得到S-扁桃酸,再经过重结晶得到纯度在99.5%以上的S-扁桃酸。S-扁桃酸的摩尔收率为78%以上(以甲苯计算)。
溴代苯类化合物在医药、农药等功能化学品的合成中起着重要作用。2,5-二溴三氟化苯是一种常用的溴代苯类化合物,它的制备方法是通过溴代反应得到。溴代反应是一种重要的化工合成反应,可以增强分子的极性,使烷烃C-H键得到活化。在溴代反应中,溴化试剂的选择非常关键。本文介绍了一种新的合成溴化剂,它是N,N-二甲基乙酰胺、氢溴酸和纯溴素的复合物。这种溴化剂具有温和、选择性高的特点,可以在一元溴化阶段停留。通过对溴代反应的工艺条件进行研究,得到了2,5-二溴三氟化苯的合成方法。
在反应瓶中加入N,N-二甲基乙酰胺、40%氢溴酸溶液,搅拌后滴加溴素,过滤得到双(二甲乙酰胺基)三溴化氢固体。然后在装有温度计和冷凝管的三口烧瓶中,加入一定量的双(二甲乙酰胺基)三溴化氢、三氟化苯和甲醇,搅拌后在室温下反应2小时。最后通过旋干、溶解、洗涤、干燥、减压除去溶剂和柱层析纯化等步骤,得到2,5-二溴三氟化苯。
在其他反应条件固定的情况下,我们考察了三氟化苯与溴化剂不同物质的量比对反应的影响。结果表明,当三氟化苯与溴化剂的量比为1∶1时,收率较高。增大投料比,收率增加不明显,考虑成本因素,选择1∶1的比例为宜。
在一定范围内,温度升高有利于反应进行。实验结果显示,在20℃时,反应的收率最高。然而,温度过高会导致副产物增多,从而降低目标产物的收率。
在上述合成条件下,我们改变反应时间,考察其对反应的影响。结果发现,反应2小时时,产率最高。延长反应时间会导致苯环溴代产物的生成,从而降低收率。
本文采用的固体溴化剂双(二甲乙酰胺基)三溴化氢具有操作简便、收率较高、副产物可回收利用、原料价格便宜等优点,比其他溴化剂更具优越性。
通过使用新型溴化剂双(二甲乙酰胺基)三溴化氢,可以高效合成2,5-二溴三氟化苯类化合物。该方法具有原料廉价易得、收率高、反应条件温和、后处理方便等优点。同时,副产物N,N-二甲基乙酰胺可以回收循环再生利用。优化的反应条件为:三氟化苯与溴化剂的量比为1∶1,反应温度为20℃,反应时间为2小时,2,5-二溴三氟化苯的收率为88%。
[1] 孙亚栋,刘方明,解正峰,等.N-(4-芳基-噻唑-2-基)-ω-(1H-苯并三唑-1-基)苯乙酮腙类衍生物的合成[J].有机化学,2005,25(4):449-453.
5-溴-3-氟吡啶是一种白色固体,常温常压下熔点为24-28度。它属于吡啶类衍生物,具有显著的碱性,可用于有机合成和医药化学中间体的制备。此外,它还可用于吡啶类配体的合成和衍生化。
5-溴-3-氟吡啶可溶于乙酸乙酯、二氯甲烷、N,N-二甲基甲酰胺和四氢呋喃等有机溶剂。它在低极性的醚类溶剂中也有一定的溶解度,但不溶于水。
5-溴-3-氟吡啶可用于有机合成和医药化学中间体的制备,特别适用于吡啶类药物分子的合成。在有机合成转化中,可以通过Suzuki偶联反应进行芳基化反应。此外,它的氟原子可被亲核试剂进攻得到脱氟官能团化的衍生物。

图1 5-溴-3-氟吡啶的应用转化
将叠氮化钠和DMF加入化学玻璃减压瓶中,然后加入3-溴-5-氟吡啶。在通风橱中,在100℃下搅拌4天。通过LC/MS监测反应混合物的完成情况。用Et2O稀释反应混合物,用水清洗反应混合物,然后在硫酸钠上干燥。通过旋转蒸发浓缩残余物,再通过柱色谱法纯化残留物,得到3-叠氮-5-溴吡啶。

图2 5-溴-3-氟吡啶的应用转化
在一个干燥的反应器中,将3'-溴苯乙酮、NBS和对甲苯磺酸溶于二氯甲烷中,然后在微波反应器中加热反应若干个小时。通过TLC点板监测反应的进度,反应结束后用二氯甲烷和水萃取所得反应混合物,分离出有机层并将其在减压下浓缩即可得到溴化的目标产物分子。
[1] Mandler, Michael D. et al Organic Letters, 24(3), 799-803; 2022
[2] Hoyt, Scott B. et al ACS Medicinal Chemistry Letters, 6(5), 573-578; 2015
正构烷烃是一种重要的化学物质,具有多种应用领域。它是一种白色晶体,不溶于水,常用于柴油、低温改进和环保液体燃料组合物的制备。此外,正构烷烃还在地质、原油、大气颗粒物和有机物等分析领域发挥着重要作用。

一种制备正构烷烃的方法如下:
(1) 将溴代正戊烷和溴代正十六烷混合形成溴代烷混合液;
(2) 称取金属钠并将其切成小细条备用;
(3) 在反应釜中加入溴代烷混合物和金属钠,升温至80℃,滴加剩余溴代烷混合液并保持微回流状,控制温度在140℃,恒温2小时;
(4) 加入乙醇和水,分离有机相,干燥后蒸馏收集正构烷烃。
该方法中,溴代正戊烷和溴代正十六烷的摩尔比为1∶1,反应釜包括搅拌器、分液漏斗和回流冷凝器,强氧化剂可选用浓硫酸或酸性高锰酸钾。
一种黑广肩步甲引诱剂的制备方法包括将β-反式罗勒烯、α-法尼烯、芳樟醇、对乙基苯乙酮、奥甘菊环、顺-3-己烯-乙酸酯、顺-3-己烯-1-醇、正十四烷、正十五烷和正二十一烷混合制备成。该引诱剂具有高灵敏度、环境友好和低成本的特点,在田间显示出良好的诱捕活性。
一种用于吸引妊娠蚊子产卵的组合物包含引诱剂和N-P-K添加剂,其中N-P-K添加剂含有氮、磷和钾的混合物。引诱剂可以调整以促进不同种类的蚊子产卵,如按蚊属蚊子、伊蚊属蚊子和库蚊属蚊子。该组合物还可以包括熟乳清、正二十一烷和/或十四烷酸甲酯。
[1] [中国发明] CN201010238781.X 一次性制备正癸烷、正二十一烷的制备方法
[2] CN201410178932.5一种黑广肩步甲引诱剂及诱芯
[3] CN201480006363.4用于引导蚊子产卵的组合物和方法
这篇文章将介绍合成和应用1-(4-甲氧苯基)-2-苄胺基丙烷的方法和相关领域的研究进展。通过对该化合物的合成及应用进行深入探讨,有望为相关领域的研究提供新的思路和方法。
简介:1-(4-甲氧苯基)-2-苄胺基丙烷是合成富马酸福莫特罗的中间体,还可合成阿福特罗等等,(R)-1-(4'-甲氧苯基)-2-苄胺基丙烷是合成(R, R)-福莫特罗的重要中间体。
1. 应用:可合成合成阿福特罗。阿福特罗是一种选择性长效β2受体激动剂,通过激活体内的腺苷酸环化酶使三磷酸腺苷(ATP)转化为环磷腺苷(cAMP),随着细胞内cAMP的增加,过敏性介质的释放 (特别是肥大细胞)被抑制,支气管平滑肌也开始松弛,从而达到缓解COPD患者症状的功效。
以成本较低的4-羟基-3-硝基苯乙酮与1-(4-甲氧苯基)-2-苄胺基丙烷为起始原料。首先,将4-羟基-3-硝基苯乙酮经非高温条件下烷基化,保护酚羟基,得到化合物3-苄氧基-4-硝基-2-苯乙酮(1),化合物1在三甲基 苯基三溴化铵的条件下(代替液溴)溴化得到潜手性3-硝基-4-苄氧基-2-溴苯乙酮(2),化合物2在工业化可购买到的催化剂RuCl[(S,S)-Tsdpen](mesitylene)的催化下,在 TritonX-100/PEG2000的双水相体系中,不对称合成(R)-3-硝基-4-苄氧基-2-溴苯乙醇(3),化合物3在温和环保条件下进行一步胺解与N甲酰化,得到(R)-N-(2-(苄氧 基)-5-(2-溴-1-羟基乙基)苯基)甲酰胺(4),化合物4经环氧化得到(R)-4-苄氧基- 3-硝基苯基环氧乙烷(5);1-(4-甲氧苯基)-2-苄胺基丙烷经手性拆分剂轻松得到(R)-1-(4-甲氧苯基)-2-苄胺基丙烷(6);化合物5与6在无溶剂加热的情况下偶联,通过一步氢化脱苄,即可得到最后的产物阿福特罗。

2. 合成:经文献检索外消旋1-(4'-甲氧苯基)-2-苄胺基丙烷的合成方法主要有三种:
2.1 方法一:以4-甲氧基苯丙酮和苄胺为原料,在Pt-C催化下高压加氢还原 得到1-(4'-甲氧基苯基)-2-苄胺基丙烷。

该方法反应条件温和,收率较高,但采用的Pt-C过于昂贵,工业化成本高。
2.2 方法二:以4-甲氧基苯丙酮为原料,利用三乙酰氧基硼氢化钠催化还原得到1-(4'-甲氧基苯基)-2-苄胺基丙烷,产率83.8%。

该方法使用的三乙酰氧基硼氢化钠制备比较繁琐,购买价格较贵,不利于大规模生产。
2.3 方法三:以4-甲氧基苯丙酮和苄胺为原料,硼氢化钠作还原剂得到对应 的盐,进而得到关键中间体1-(4'-甲氧苯基)-2-苄胺基丙烷。

3. (R)-1-(4'-甲氧苯基)-2-苄胺基丙烷的合成:由外消旋1-(4'-甲氧苯基)-2-苄胺基丙烷得到(R)-1-(4 '-甲氧基苯基)-2-苄胺基丙烷主要的合成方法就是利用(S)-扁桃酸拆分得到相应的(R)-1-(4'-甲氧苯基)-2-苄胺基丙烷-(S)-扁桃酸盐,再在碱性条件下得到游离胺(R)-1-(4'-甲氧苯基)-2-苄胺基丙烷,ee值可达99.5%。

参考文献:
[1]罗茂. (R,R)-福莫特罗及其中间体的合成研究[D].浙江理工大学,2023.DOI:10.27786/d.cnki.gzjlg.2023.001193.
[2]王宁. 汇聚式策略制备阿福特罗的新工艺研究与优化[D].河北大学,2021.DOI:10.27103/d.cnki.ghebu.2021.001670.
本文旨在介绍合成对乙基苯乙炔的方法和策略,以帮助读者了解如何有效地合成这一关键化合物。
背景:液晶显示器的响应时间是其重要的性能指标之一,而具有大阈值(△n)和低粘度(η)的液晶材料是实现快速响应液晶显示器的关键条件。二苯乙炔类液晶材料具有大阈值和低黏度,能够改善混合液晶的双折射率和清凉点,降低液晶的黏度,提高液晶的响应速度。因此,它们常被用作高级TN、STN和TFT液晶显示器中的混晶配方的稀释剂。因此,合成二苯乙炔类液晶材料具有很大的实用价值。采用以碘代烷基苯(或联苯)(I)与烷基苯乙炔(Ⅱ)为中间体,利用二价镍催化剂通过偶联反应一步合成目标化合物,可以显著降低成本。因此,探讨作为中间体的4-乙基苯乙炔的合成是非常必要的。
合成:
以乙苯作为原料,通过乙酰化、还原、脱水、溴化和消除五步反应合成了对乙基苯乙炔。具体步骤如下:
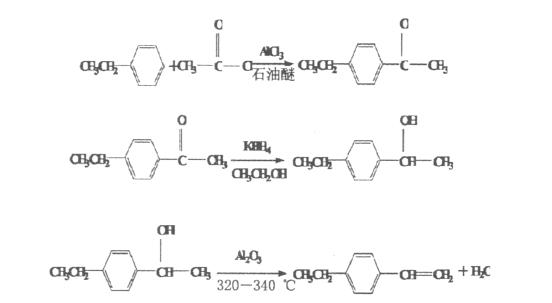
(1)乙基苯乙酮的合成
在1000毫升的三口瓶中加入106克乙基苯、147克三氯化铝和250毫升石油醚,搅拌并降温至10.0摄氏度,然后滴加乙酰氯,控制温度低于10摄氏度,加完后,让其在室温下反应2小时。接着慢慢滴加80毫升盐酸和535毫升水,控制温度低于20摄氏度,加完后搅拌均匀,然后进行分液。将水相用100毫升二氯甲烷进行两次提取,合并有机相,用100毫升水进行两次洗涤,回收二氯甲烷和石油醚,然后通过减压得到118克产品(GC>98%),收率为79.73%。需要注意的是,本反应中乙基苯最好不要反应不完全,产品中不能含有大量杂质。
(2)乙基苯乙醇的合成
1000 mL三口瓶中加入148 g乙基苯乙酮,280 mL无水乙醇,搅拌,滴加18 g KBH4及150 g水的混合物,约30-40 min加完,室温下反应2 h;然后慢慢滴加33 mL盐酸,反应放热并升温至有回流,加毕,室温下反应2 h;然后加入130 mL二氯甲烷搅匀,静置,分层,二氯甲烷相水洗3次(不如改为60- 90℃石油醚方便操作),回收二氯甲烷,减压得无色透明液体,产量为120 g(GC>96%),收率80%。其中的杂质主要是已脱水的产物,不影响后续步骤的反应 质量。
(3)乙基苯乙烯的合成
1000 mL三口瓶中加入750.0 g乙基苯乙醇,安装三氧化二铝电加热柱,冷凝器,水抽泵,使水抽泵真空0.09-0.1 MPa,三氧化二铝填充柱电加热使达到320-340℃,1000 mL三口瓶电加热,约6-7 h蒸完(脱水不能太快,否则脱水效率不好),蒸出物分去下层水,上层为乙基苯乙烯粗品,经硫酸钠干燥 24 h,过滤除去硫酸钠,滤液水洗加热减压蒸馏得:64℃/399.9 Pa产物552 g(GC>99%),收率 80.0%。
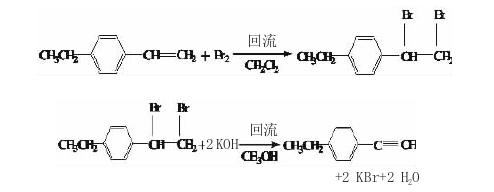
(4)乙基苯二溴乙烷的合成
1000 m L三口瓶中加入乙基苯乙烯138.0 g, 420 mL二氯甲烷,搅拌,滴加167.0 g溴,保持微回流,加毕,回流反应5-10 min,滴加200 mL水,加入 8.0 g亚硫酸氢钠固体,分液,水洗两次,硫酸钠干燥 24 h,回收二氯甲烷,剩余物加入2倍即550 mL乙醇,搅匀,冷却,抽滤,得白色固体274 g(GC≥99.0%),收率:92%。
(5)对乙基苯乙炔的合成
1000 m L三口瓶中加入乙基苯二溴乙烷298.0 g和 255 mL甲醇,搅拌,分批加入氢氧化钾343.0 g,反应放热,加毕,回流反应6 h,停止加热,将物料倒入 500.0 m L水中,补入250 mL石油醚,搅匀,分液,醚层水洗两次,硫酸钠干燥24 h,回收石油醚,剩余物减压得57-59℃/666.5 Pa产物,残液中乙基苯溴乙烯加入100 g氢氧化钾,250 mL甲苯,回流反应 2 h,加入250 mL水中,分层,水洗甲苯层,硫酸钠干燥,回收甲苯,余液减压蒸馏又得苯乙炔,总计得 121.0 g(GC>99.0%),收率为88.97%。
参考文献:
[1]陈颖.乙基苯乙炔的合成[J].河北化工,2007,(08):35-36+39.
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手

























