溶剂兰97是一种取代的氨基蒽醌,可用于塑料和合成纤维的染料,以及羊毛染料的前体。

在氮气下,将醌茜、二氢醌茜、硼酸三甲酯和乳酸引入2,6-二乙基-4-甲基苯胺中,经过一系列加热和蒸发的步骤,得到溶剂兰97。
溶剂兰97可用于制备高色彩对比度眼镜镜片,其中包括基片和强化膜层。基片由聚酰胺或聚碳酸酯制成,分散剂和溶剂的混合物用于调制基片,紫外线吸收剂用于增加眼镜的功能。强化膜层通过浸泡镜片于液态硅化物中并经过热烤干燥而形成。
[1]CN200710194781.2取代的氨基蒽醌的制备
[2]CN201410232430.6高色彩对比度眼镜镜片及制备方法
异兰,异索兰是一种存在于多种植物中的化合物,主要分布在Primula属植物中。它具有抗增殖活性,对M109鼠癌细胞和A2780人卵巢癌细胞系具有抑制作用(IC50¼10mg/mL和IC50¼2.9mg/mL)。此外,它还表现出对链球菌的抗菌活性。


制备方法如下:
[1] Sisa M , Dvorakova M , Vanek T . Concise access to primin, miconidin and related natural resorcinols[J]. Tetrahedron, 2017, 73(35).
磷酸二苄酯是一种重要的有机磷酸酯类化合物。具有分子式C??H??O?P,分子量为278.24。磷酸二苄酯是一种白色至微黄色的粉末状物质,在常温常压下相对稳定。需要严格的储存条件,保持在2-8°C的低温环境中,以防止变质或分解。该化合物微溶于水,但可溶于甲醇等有机溶剂,这为其在化学反应和应用中的溶解性提供了便利。磷酸二苄酯在药物合成、化学研究及材料科学等领域发挥着重要作用,具有高纯度和良好的化学稳定性。

磷酸二苄酯的性状
磷酸二苄酯的合成方法包括苯甲醇、三氯化磷和三乙胺在甲苯中混合加热回流反应。这一步骤生成亚磷酸二苄酯,纯度约为69%。随后的反应步骤进一步提高了磷酸二苄酯的纯度至97%以上。最终通过精制过程将其纯度提高至99.5%以上。
磷酸二苄酯作为医药中间体在药物合成中发挥着重要作用。除此之外,它还被广泛应用于化学试剂和精细化学品的制备中。在实验室中,磷酸二苄酯常被用于合成其他有机化合物、催化反应以及作为分析试剂等。
[1] 魏彦君,刘希望,王文才,等.磷酸二苄酯及焦磷酸四苄酯的制备方法:CN202011138934.3[P].CN202011138934.3[2024-09-03].
[2] 支海兰.磷酸二苄酯在稀土转型和回收中的应用研究[D].福建师范大学,2021.
[3] 王勇,张喜全,郭猛,等.磷酸二苄酯及其衍生物的制备方法:201910084365[P][2024-09-03].
氧肟酸是一类常见的脲酶抑制剂,为国外七十年代开发的用于尿结石及尿道感染的药物。其中,乙酰氧肟酸(Acetohydroxamicacid,AHA)被认为是氧肟酸类化合物中最有效的一种脲酶抑制剂,可由化学合成法制得。
由于乙酰氧肟酸具有-CONHOH 功能团,在医药上,可与尿素酶的镍原子螫合,使尿素酶失活,从而阻止尿素被分解,恢复尿液正常酸碱度,改善磷酸铵镁和含碳酸盐的磷灰石过饱和的生理环境,防止结石的形成及抑制其长大。又由于AHA具有弱酸性,可使结石逐渐溶解、缩小甚至消失。AHA又有一定的抗菌活性,可用于预防及治疗尿路插管结石、尿路结石。在农业上,乙酰氧肟酸还可作为脲酶竞争性抑制剂,减缓尿素或降低蛋白质分解为氨的速度,从而提高蛋白质的利用率.
利用羟胺在碱性条件下活性最强的特点,将乙酸乙酯或乙酸甲酯与羟胺在碱性溶剂体系中合成AHA,用到的原理是酰胺化反应。

副反应有羟胺的分解和乙酸酯的水解。
以羟胺为亲核试剂,在显弱碱性的醇溶剂中,与醋酸甲酯发生反应得到乙酰氧肟酸,然后通过滴加强碱,反应生成乙酰氧肟酸盐,从而使该可逆反应正向进行,得到大量的乙酰氧肟酸盐。
苏兰辉等人采用乙酸乙酯和盐酸羟胺在以甲醇为溶剂的体系中反应制取乙酰氧肟酸,探讨了乙酰氧肟酸的合成工艺条件.
在四口反应瓶中加入甲醇,水,盐酸羟胺,再加入乙酸乙酯和NaOH溶液,控制一定温度,使其充分反应一段时间,最后加入浓盐酸使NaCl沉淀从体系中析出。将反应混合物真空浓缩后,用沸腾的乙酸乙酯萃取,向萃取液中滴加几滴丙酮,使未完全反应的盐酸羟胺转化为丙酮肟,浓缩萃取液,冷却母液,析出大量的晶体,于65℃真空干燥箱中干燥4h,得到成品。
利用盐酸羟胺与乙酸乙酯反应可合成出AHA,较佳的工艺条件是:盐酸羟胺:乙酸乙酯=1:1.1,反应时间为2h,反应温度为27-33℃,所得乙酰氧肟酸产率84%,纯度97%。碱性的甲醇水溶液体系,当其用量为乙酸乙酯的2.3倍(体积分数)时,产品的产率及纯度均较好。
刘蔷以聚乙烯醇解液、盐酸羟胺为原料,经过酰胺化反应制备乙酰氧肟酸,并采用正交实验和单因素实验对中间体乙酰氧肟酸的制备进行了优化。产物收率为95.3%,经HPLC 检测纯度为98.1%.
[1] 王淑香,闫明伟.乙酰氧肟酸(AHA)型脲酶抑制剂提高奶牛产奶量的理论与实践[J]. 饲料博览, 2004(7): 46.
[2] 钟祥,梁娟秋.新型螯合萃取剂.金属学报, 1982, 18(2): 221-225.
[3] 黄旋林,吴祥林.异肟酸类型捕收剂的研究与浮选稀土矿物实验.稀土, 1985, 16(3): 1-4.
[4] 刘成梅,游海.天然产物有效成分的分离与应用[M].北京:化学工业出版社,2003,174.
[5] 苏兰辉,江向阳. 乙酰氧肟酸合成工艺条件探讨[J]. 中国饲料,2005, 15(14): 15-16.
[6] 刘蔷. PVA 醇解液制备乙酰氧肟酸和苄氧胺盐酸盐新工艺研究[D]. 太原:中北大学, 2014.
制备氯化石蜡是有机合成中的重要步骤,其方法多样且具有一定的技术挑战性。通过合适的实验条件和反应控制,可以高效地合成出这种化合物。
简述:氯化石蜡又称氯烃,是C10~C 30石蜡烃的氯化衍生物。根据含 氯量的不同主要有氯化石蜡-42、氯化石蜡-52、氯化石蜡-70产品。氯化石蜡又挥发性低、阻燃、电绝缘性良好,广泛应用于聚氯乙烯的增塑剂,特别是氯乙烯—醋酸乙烯共聚树脂可单独使用,同时还可作橡胶、涂料、油墨、油漆、聚氨酯发泡添加剂、阻燃剂和各种切削油、润滑油的耐极压添加剂等。
制备:
(1)制备
按照下图所示将回流反应装置、干燥装置、碱液吸收装置和抽真空惰性气体置换保护装置依次连接。在氩气保护下,用注射器将 1 mL 烷烃、30 mL 二氯甲烷、9 mL 磺酰氯通过翻口塞注入两口反应瓶中,打开汞灯柱( 10 W) 光照,70 ℃搅拌加热回流反应。达到反应时间后冰水浴冷却终止反应,打开反应瓶的翻口塞,通氩气吹扫反应体系 10 min,将反应溶液转移到旋蒸瓶中,50 ℃ 下减压旋蒸,蒸出二氯甲烷和过量的磺酰氯。冷却至室温后,向残留物中加入 40 mL 二氯甲烷,用 40 mL 饱和氯化铵水溶液洗涤 3 次、40 mL 饱和碳酸氢钠水溶液洗涤 3 次以及 40 mL 去离子水洗涤 3 次至水相呈中性,然后用 40 mL 饱和食盐水洗涤。有机相用无水硫酸钠干燥、过滤、减压下除去溶剂,50 ℃真空干燥后得产物。

(2)分离纯化
取200 μL干燥产物,用正己烷稀释至6 mL。取1 mL稀释溶液上柱(柱尺寸为300 mm×10.5 mm),从下往上依次装填3 g弗罗里硅土、2 g中性硅胶、5 g32%酸性硅胶、4 g无水硫酸钠。首先用50 mL正己烷洗涤柱子,丢弃洗涤溶液,然后用100 mL的V(二氯甲烷):V(正己烷) = 1:1的溶液洗脱。在50 ℃下减压蒸馏去除溶剂,最终得到高纯度氯化石蜡产物。
参考文献:
[1]张大景. 氯化石蜡-52生产自动化工艺分析 [J]. 中国氯碱, 2023, (09): 29-32.
[2]李慧娟,刘兰畦,赵梅等. 不同氯化度的氯化石蜡制备方法研究 [J]. 山东科学, 2021, 34 (05): 97-103.
[3]闫国强. 光催化法生产氯化石蜡-52工艺研究 [J]. 广东化工, 2017, 44 (14): 74-75.
本文将探讨合成2-氨基-5-甲氧基吡啶的方法,这是一种重要的医药中间体,在药物合成领域具有广泛应用。
背景:2-氨基-5-甲氧基吡啶类化合物是一类极为重要的医药中间体,其衍生物在治疗多种疾病方面具有显著疗效。例如,吡罗昔康是一种高效的消炎药,对关节炎有特殊疗效;烟乙酰胆碱类化合物在阿尔茨海默氏痴呆症、抑郁症、帕金森症、止痛、精神分裂症等疾病的治疗中展现出良好的效果。
合成:
2-氨基-5-甲氧基吡啶的合成比较困难,在James A.Moore和Frank J的方法中,乙醇在密封管中加热到了150℃,条件比较苛刻。而Joseph G.ombardino的方法比较温和,兰天等人在此基础上对反应进行了优化。具体步骤如下:
(1)2-溴-3-羟基吡啶的制备
用冰盐浴把含160克(1mol)Na0H的600ml水溶液冷至-10-0℃,保持温度滴加159.8克(1mo1)液溴。将97.9克(1mol)3-羟基啶溶于含40克(1mol)Na0H的500ml水溶液中,并把此溶液滴入上述的溴溶液中,保持体系温度15℃。滴加完成后,室温搅拌3小时。用浓硫酸酸化体系至pH=7,伴随着有大量白色产物出现,如果不能有效搅拌,则先把产物抽滤,滤液再进行酸化。所得粗产物用70%乙醇重结晶,得无色晶体,mp184-186℃,产率:135g(78%)。
(2)2-溴-3-甲氧基吡啶的制备
将7克的钠小心的加入200ml甲醇中,保持体系温和回流,接着将溶有50克2-溴-3-羟基吡啶的500mLDMF溶液加入其中。搅拌片刻后用水泵减压蒸出大量甲醇(18mm/Hg),余下的混合物中加入18.8ml甲烷,室温搅拌过夜,然后用油泵减压蒸出DMF(10mm/Hg),冷至室温后得到深黄色固体,加入乙醚多次萃取,分出有机相,并用饱和食盐水洗两次,有机相用无水MgS04干燥蒸馏的黄色结晶产物,mp43-45℃,产率:43g(80%)。
(3)2-溴-3-甲氧基-5-硝基吡啶的制备
在0℃时把40克2-溴-3-甲氧基吡啶加入到剧烈搅拌的160ml浓硫酸和160m1发烟硝酸混合液中,用时40分钟。接着50-55℃加热反应1小时,冷却后将其滴入4000m1冰水中,边滴边搅拌。析出大量浅黄色固体,抽滤,干燥,得到浅黄色针状晶体,mp 134-135℃,产率:40g(80%)。
(4)2-氨基-5-甲氧基吡啶的制备
将200毫升10% Pd/C和0.394克NaOH溶解于400毫升乙醇中,随后加入2.0克2-溴-3-甲氧基-5-硝基吡啶。在常压条件下进行催化加氢,当吸收4当量的氢气后停止反应。经抽滤后,滤液经6当量盐酸酸化处理,并通过旋转蒸发仪除去溶剂。随后,用10mol/L的NaOH碱处理混合物,再用乙醚苯进行萃取,萃取液经无水硫酸钠干燥。蒸发溶剂后,通过柱色谱纯化得到0.62克红色粘稠液体,产率为59%。
参考文献:
[1]兰天.面不对称二茂铁衍生物的合成研究[D].兰州大学,2006.
本文将介绍合成头孢他啶侧链酸活性硫酯的具体步骤和操作技巧,通过深入探讨合成过程中的关键因素,旨在为读者提供合成头孢他啶侧链酸活性硫酯的指导和参考。
背景:头孢他啶(Ceftazidine)于1978年被发现, 1983年英国葛兰素公司首先开发上市,1992年在中国首次上市,1993年被正式列入中国国家基本 药物目录。20多年的临床应用验证,头孢他啶具有抗菌谱广、耐酶等特点,它对铜绿假单胞菌的抗菌活性较强,被广泛用于治疗各种中、重度感染,是治疗感染革兰阴性菌危重患者的首选药物。
头孢他啶侧链酸活性硫酯,其化学名称为2-(2-氨基-4-噻唑基)-2-(叔丁氧羰基)-异丙氧亚氨基乙酸硫代苯并噻唑酯,它不仅是合成头孢他啶的重要中间体。而且是合成新型头孢菌素包括新型脒基吡啶基头孢菌素类化合物、含有环丙烷并氮杂环的头孢菌素衍生物、氨曲南等的重要原料。头孢他啶侧链酸活性硫酯的品质和收率直接影响所合成头孢他啶及新型头孢菌素的质量好坏和成本。
合成:
往往以头孢他啶侧链酸为原料,在有机碱作催化剂条件下,与DM反应,生成头孢他啶侧链酸活性酯。
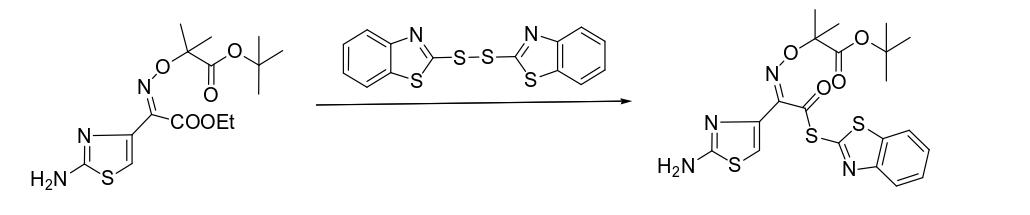
1. 方法一:
向装有冷凝管和机械搅拌装置的四口瓶中依次加入三苯基膦(20.5 g、0.078 mol)、精制的DM(25 g、0.078 mol)、三乙胺(6.03 g、0.06 mol)于75 mL甲苯和75 mL二氯甲烷的混合液中,搅拌1 h.在2 h内分批加入头孢他啶侧链酸(19.8 g、0.06 mol),控制温度不超过5 ℃, 加完后搅拌4 h,将反应液冷冻,抽滤,分别用甲苯50 mL和乙醚30 mL洗涤,60 ℃下真空干燥,得到浅黄色固体16.6 g,收率为75%.产品mp为134 ℃~136 ℃,Rf=0.42(硅胶板,展开剂PE∶AcOEt=3∶1)。
2. 方法二:
头孢他啶侧链酸(19.8 g、0.06 mol),乙腈150 mL,混合后搅拌约30 min,加入三乙胺(7.84 g、0.078 mol)、DM(25 g、0.078 mol)于混合液中搅拌.降温至5 ℃,滴加亚磷酸三乙酯(12.9 g、0.078 mol),约1 h滴加完毕,继续反应4 h,抽滤,固体用乙醚洗至浅黄色,真空干燥,得头孢他啶活性硫酯18.08 g,收率86.8%.产品为淡黄色固体,产品mp:134 ℃~136 ℃,Rf=0.42(硅胶板,展开剂PE∶AcOEt=3∶1)。
3. 方法三:
傅德才等可采用二氯甲烷及甲苯混合物作为反应溶剂,在碱性条件下,他啶酸与DM、三苯基膦反应,反应温度≤30 ℃,反应时间8 h,收率可达79.6%。 此法的缺点是甲苯与二氯甲烷的混合溶剂中,夹杂有少量未反应的头孢他啶侧链酸,不易于产物的纯化。
4. 方法四:
张平等以头孢他啶侧链酸为原料,甲苯为溶剂,三乙胺为催化剂,与三苯基膦、二硫二苯并噻唑(DM)反应,反应温度25 ℃,反应4 h,收率为85.4%, 纯度99.3%。
5. 方法五:
哈药集团制药总厂的王庆全等以头孢他啶侧链酸为原料,二氯甲烷为溶剂,三乙胺为催化剂,反应温度35 ℃,与三苯基膦、二硫二苯并噻唑反应3 h, 收率可达90.17%,含量为97-98%。此工艺缺点是以二氯甲烷为溶剂,部分产品溶于其中,影响收率。这三种方法都使用了三苯基膦,由于三苯基膦价格昂贵, 造成生产成本较高。
6. 方法六:
美国专利US 4652651,US 4695639提到在乙腈中,采用甲基吗啉为催化剂, 他啶酸与DM、亚磷酸三乙酯在0 ℃反应5 h,收率可达76.5%-80.3%。
7. 方法七:
梁宝臣等以乙腈为溶剂,反应温度≤30 ℃,在碱性条件下,他啶酸与DM、 亚磷酸三乙酯反应约7h,收率为81%。
参考文献:
[1]厉昆,李啸风,任红阳等. 头孢他啶侧链活性酯中一个未知杂质的合成和确认 [J]. 精细化工中间体, 2015, 45 (01): 57-60+68. DOI:10.19342/j.cnki.issn.1009-9212.2015.01.017
[2]杨晓辉. 头孢他啶侧链酸及其活性酯的合成研究[D]. 天津大学, 2012.
[3]王玉环. 头孢他啶侧链酸活性硫酯的合成研究 [J]. 石家庄职业技术学院学报, 2009, 21 (02): 33-35.
[4]傅德才,冀学时,李忠民. 头孢他啶侧链酸活性硫酯的合成 [J]. 中国医药工业杂志, 2002, (11): 5-6.
本文旨在探讨合成2'4'-二氟-2-[1-(1H-1,2,4-三唑基)]苯乙酮的方法,通过本文的研究,将为2'4'-二氟-2-[1-(1H-1,2,4-三唑基)]苯乙酮的生产提供新的技术支持和方法。
背景:2'4'-二氟-2-[1-(1H-1,2,4-三唑基)]苯乙酮是合成氟康唑的重要中间体。
氟康唑是一种新型的三唑类抗真菌药物,具有制菌谱广、口服吸收完全、组织器官内浓度高等特点。此外,它还具有毒副作用小、临床效果好等优点。氟康唑的作用机理是通过抑制细胞内细胞色素P450催化的羟化反应,从而阻止麦角固醇合成,破坏真菌细胞的完整性,阻止真菌细胞的生长和繁殖。因此,氟康唑是深部抗真菌的首选药物。然而,由于其价格一直较高,人们一直致力于降低其生产成本,对氟康唑及其中间体进行研究。
合成:
1. 方法一:
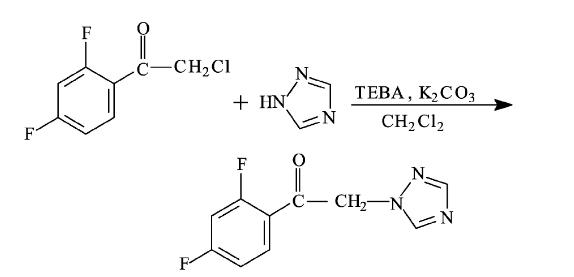
(1)在装有温度计、电动搅拌器、恒压滴液漏斗和回流冷凝管的500 mL四口烧瓶中,依次加入1,2,4-三氮唑8.97 g(0.13 mol)、三乙基苄基氯化铵(TEBA) 0.5 g (2.70 mmol,1,2,4-三氮唑摩尔用量的2%)、碳酸钾13.8 g(0.1 mol)及二氯甲烷50 mL。开动搅拌器,将四口烧瓶置于0℃的冰水浴中,开始滴加溶于50 mL二氯甲烷的2-氯-2′,4′-二氟苯乙酮19.05 g(0.1 mol),滴加完毕后于常温(25~30℃)反应6 h,然后过滤残渣,减压回收溶剂,得粗品22.1 g。
(2)将粗品倒入50 mL冰水中,加入稀盐酸,使粗品全部溶解,静置分层后,用分液漏斗除去不溶油状物,水层用碳酸氢钠中和,减压过滤,可获得白色结晶18.4 g,相对分子质量为223.18,熔点104~105℃。
2. 方法二:
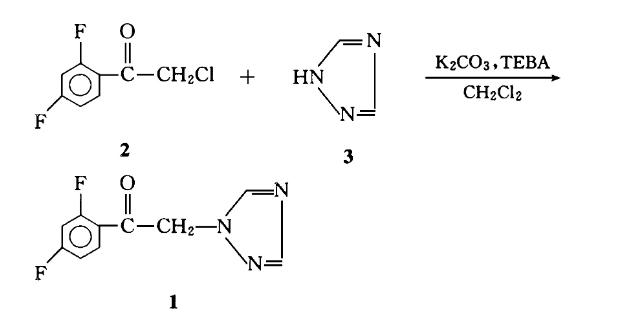
将1H-1, 2, 4-三唑 (3) (8.97 g,0.13 mol) 、TEBA (0.5 g ,2.70 mmol) 、K2CO3 (13.8 g,0.13 mol) 悬浮于CH2Cl2 (50 ml) 中,于0°C滴加2 (19.05 g,0.1 mol) 溶于CH2Cl2 (50 ml) 的溶液,滴毕于常温反应6h,然后滤除残渣,减压回收溶剂,即得粗品 (22.1g) 。将粗品倒入冰水 (500ml)中,加入1mol/L盐酸 (120ml) 将粗品全部溶解,静置分层除去不溶油状物,水层用NaHCO3 (8g) 中和至pH6,得白色沉淀2'4'-二氟-2-[1-(1H-1,2,4-三唑基)]苯乙酮 (18.4 g,82.6%) ,mp 105~106℃。
参考文献:
[1]常瑜,翟红,王秀兰. 2′,4′-二氟-2-(1H-1,2,4-三唑-1-基)苯乙酮的合成 [J]. 太原理工大学学报, 2006, (06): 615-617. DOI:10.16355/j.cnki.issn1007-9432tyut.2006.06.004
[2]钟武,张万年,李科等. 2′,4′-二氟-α-(1H-1,2,4-三唑-1-基)苯乙酮的合成 [J]. 中国医药工业杂志, 1999, (09): 36. DOI:10.16522/j.cnki.cjph.1999.09.017
硫酸阿巴卡韦是一种重要的抗病毒药物,其合成方法对于提高药物的生产效率和质量具有重要意义。在本文中,我们将探讨如何通过相关的合成路径来制备硫酸阿巴卡韦。
背景:阿巴卡韦是一种抗艾滋病新药,最早由英国葛兰素韦康公司生产,为新的碳环2’-脱氧鸟苷核苷类药物,其口服生物利用性高,易深入中枢神经系统。1988年美国批准硫酸阿巴卡韦的片剂和口服液上市,已在世界28个国家地区上市销售,硫酸阿巴卡韦片剂及口服液2000年在中国授权,2002年上市销售原有药品行政保护,但于2007年10月28日到期。阿巴卡韦片剂只需早晚各服用一片就能达到之前抗艾滋病其他药物的几十片的疗效。
合成:
以(1S, 4R)-4-氨基-2-环戊烯-1-甲醇和 2-氨基-4,6-二氯-5-甲酰胺基嘧啶为起始原料经过缩合反应、环合反应、亲核取代反应和成盐反应,最终得到硫酸阿巴卡韦,收率 90%左右,纯度为 99.66%。具体实验步骤如下:
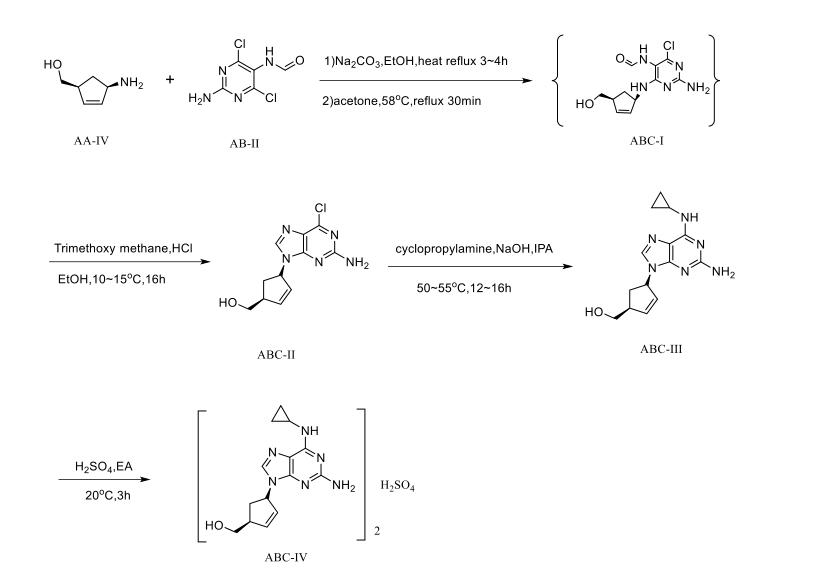
(1)化合物 ABC-II((1S,4R)-cis-4-[2-氨基-6-氯-9H-嘌呤-9-基]-2-环戊烯-1-甲醇)的合成
称取AA-V(11.97 g)置于250 mL三口瓶中, 在N2保护下溶于乙醇(300 mL)中,待 AA-V 完全溶解后,加入碳酸钠(18.3 g),继续搅拌 30 min,然后加入AB-Ⅰ(14.91 g),加热回流 3~4 h。HPLC 监测 AB-Ⅰ<3% 减压蒸除溶剂,剩余物中加入丙酮(190 mL),在 58℃下回流 30 min,降低温度至 30℃,抽滤,滤液减压蒸除溶剂,蒸干得黄色泡状物 ABC-I(22.84 g)。称取 ABC-I(22.84 g)置于 1 L 的三口烧瓶中,加入乙醇(145 mL)、原甲酸三乙酯(245 mL)以及 35% HCl 溶液(14.5 mL),控制温度在 10~15℃ 之间搅拌 16 h。抽滤,滤饼用 30 mL 乙醇打浆洗涤两次,干燥,得到产品 ABC-II (18.51g)。
(2)化合物 ABC-III((1S,4R)-4-[2-氨基-6-(环丙胺基)-9H-嘌呤-9-基]-2-环戊烯-1-甲醇)的合成
称取 ABC-II((1S, 4R)-cis-4-[2-氨基-6-氯-9H-嘌呤-9-基] -2-环戊烯-1-甲醇)(18.51 g)置于 100 mL 三口烧瓶中,然后加入异丙醇(124 mL)和环丙胺(62mL),控制温度在 50~55℃ 之间加热 12-16 h。HPLC 监测 ABC-II<1%,降低温度至室温,加入等当量的 4 mol/L NaOH 溶液,减压旋蒸蒸除溶剂,加入异丙醇(60 mL),抽滤,保留滤液。浓缩滤液,得到油状物,加入丙酮(124 mL)处理得到泥浆状物。抽滤,丙酮洗涤,干燥,得 ABC-III 产品(15.01 g,易潮解,室温减压抽干得浅黄色固体)。
(3)化合物 ABC-IV(硫酸阿巴卡韦)的合成
称取 ABC-III (15.01 g)置于 100 mL 单口瓶中,加入乙酸乙酯(75 mL),滴加浓硫酸的乙酸乙酯溶液(1.5 mL 浓硫酸 + 25 mL 乙酸乙酯),滴加完毕之后,控制温度 20℃ 保温搅拌 3 h,抽滤,少许 EA 洗涤,抽干后,乙醇打浆洗涤,烘干,得到白色产品 ABC- IV(13.49 g)。
参考文献:
[1]岳祥军, 一种氨基醇的拆分方法. 安徽省, 安徽贝克联合制药有限公司, 2017-05-17.
[2]李国俊. 抗艾滋病药物硫酸阿巴卡韦的合成及其工艺研究[D]. 安徽中医药大学, 2017.
合成8-(苄氧基)-5-(2-溴乙酰基)喹诺酮是一项关键的有机合成研究,具有重要的药物学意义。
背景:β2肾上腺素受体激动剂是治疗哮喘和慢性阻塞性肺疾病的主要药物之一。目前,临床上常用的抗哮喘药物包括茚达特罗、卡莫特罗、LAS100977等,这些药物的合成过程通常涉及关键中间体8-(苄氧基)-5-(2-溴乙酰基)喹诺酮(Ⅰ)与相应的胺缩合、脱保护基团等步骤。
合成:
以8-羟基喹啉-N-氧化物为起始原料,经转位、Fries重排、苄基化和溴化等反应合成了抗哮喘药中间体8-苄氧基-5-(2-溴乙酰基)喹诺酮。具体实验步骤如下:

(1)8-乙酰氧基喹诺酮 (Ⅲ) 的合成
2.50kgⅡ与12kg乙酰酐加到50升搪瓷反应釜中搅拌、加热至122℃回流反应4h后冷却。反应完全后夹套通冷却水降温。物料放入离心机甩干,并用乙酸酐浇洗后再甩干,滤液回收备用,得湿料2.8kg,在80℃下烘干,得2.5kg白色固体Ⅲ。含量 (HPLC) 98.5%,收率78.2%,m.p.243.2~245.0℃。
(2)5-乙酰基-8-羟基喹诺酮 (Ⅳ) 的合成
在0℃,2.5kgⅢ与3.8kg无水AlCl3和75g乙酰氯混合在55kg二氯乙烷中,混合物加热到75~85℃反应4h,减压蒸馏除去70%~80%溶剂,然后快速地加入12kg水,再用5kg 30%HCl处理。冷却到10℃,搅拌3h,接着用离心过滤,滤饼用冷水洗涤,接着在80~95℃真空下干燥。2.5kgⅣ的粗品溶解在27kg二氯甲烷中,混合物加热到回流并过滤掉不溶物。滤液冷却到0℃,过滤出产品并用二氯甲烷洗涤。母液再与不溶物于30℃时混合搅拌12h,过滤除去不溶物,滤液再冷却,过滤出第二次产物并用二氯甲烷洗涤,合并滤饼,在80℃下干燥6h,得2.4kg黄绿色固体Ⅳ。纯度96.81%,收率94.7%。m.p.>260℃ (变黑分解) 。
(3)5-乙酰基-8-苄氧基喹诺酮 (Ⅴ) 的合成
在-2℃条件下,将2.4千克Ⅳ与2.0千克碳酸钾和10克KI混合于20千克DMF中,然后在6小时内将1.5千克氯化苄滴加至反应釜中。混合物在15~20℃搅拌至反应完成,然后减压蒸馏去除80%的DMF,随后用2×30千克饱和食盐水洗涤。进行抽滤,将滤饼溶解于二氯甲烷,经无水硫酸镁干燥,再经硅藻土过滤,将滤液减压浓缩至得到3.1千克淡黄褐色粉末Ⅴ。其纯度为97.63%,收率为90.1%,熔点为169.8~171.5℃。
(4)8-苄氧基-5- (2-溴乙酰基) 喹诺酮 (Ⅰ) 的合成
3.1kgⅤ溶解在42kg二氯甲烷中,缓慢加入1.8kg三氟化硼乙醚复合物,加热到30~35℃,慢慢滴加1.9kg液溴和10kg二氯甲烷的混合物,加完后回流1h,在2h内蒸馏出溶剂然后冷却到10~15℃。加入碳酸钾水溶液,过滤并用水洗涤,在低于95℃下干燥12h。用12kg甲醇与氯仿混合溶液 (体积比1∶1) 搅拌研磨1h,抽滤,干燥得3.3kg灰白色粉末Ⅰ。纯度98.40%,收率84.6%。m.p.195.7~196.2℃,用二氧六环重结晶,熔点提高到201~204℃。
参考文献:
[1]李树安,谭超兰,张丹丹等.8-苄氧基-5-(2-溴乙酰基)喹诺酮的合成[J].精细石油化工,2018,35(02):40-43.
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手
























