| 中文名称 | 灭菌唑 | 英文名称 | triticonazole |
|---|---|---|---|
| 中文别名 | (E)-5-(4-氯亚苄基)-2,2-二甲基-1-(1H-1,2,4-三唑-1-基甲基)环戊醇;(RS)-(E)-5-(4-氯苯亚甲基)-2,2-二甲基-1-(1H-1,2,4-三唑-1-基甲基)环戊醇;(±)-(E)-5-(4-氯苄烯基)-2,2-二甲基-1-(1H-1,2,4-三唑-1-基甲基)环戊醇;扑力猛; |
英文别名 | Triticonazole;(RS)-(E)-5-(4-chlorobenzylidene)-2,2-dimethyl-1-(1H-1,2,4-triazol-1-ylmethyl)cyclopentanol;(5E)-5-[(4-chlorophenyl)methylene]-2,2-dimethyl-1-(1H-1,2,4-triazol-1-ylmethyl)cyclopentanol;(5Z)-5-[(4-chlorophenyl)methylidene]-2,2-dimethyl-1-(1,2,4-triazol-1-ylmethyl)cyclopentan-1-ol;rac-(1R,5E)-5-[(4-chlorophenyl)methylidene]-2,2-dimethyl-1-(1H-1,2,4-triazol-1-ylmethyl)cyclopentan-1-ol; |
| CAS号 | 131983-72-7 | 分子式 | C17H20ClN3O |
| 分子量 | 317.81300 | 精确质量 | 317.12900 |
| PSA | 50.94000 | LOGP | 3.56620 |
灭菌唑对大鼠急性经口LD50>2000mg/kg,大鼠急性经皮LD50>2000mg/kg,大鼠急性吸入LC50(4h)>1.4mg/L。对兔眼睛和皮肤无刺激。山齿鹑急性经口LD50>2000mg/kg,虹鳟鱼LC50(96h)>10mg/L,水蚤LC50(48h)>9.3mg/L,对蚯蚓无毒。
【原 药】 95%
灭菌唑有25克/升悬浮种衣剂,300克/升悬浮种衣剂两种剂型。
灭菌唑是甾醇生物合成中C-14脱甲基化酶抑制剂,主要用作种子处理剂,也可茎叶喷雾,持效期长达4~6周。适用于禾谷类作物、豆科作物、果树病害,对种传病害有特效。
【对作物安全性】 推荐剂量下对作物安全、无药害。
灭菌唑主要用于防治禾谷类作物如小麦、大麦、油菜、花生、水稻和豆类作物等众多病害。对所有麦类病害都有很好的防治效果,如小麦和大麦的白粉病、纹枯病、枯萎病、叶斑病、锈病、菌核病、网斑病、云纹病等。还能防治油莱和花生的土传病害,如菌核病,以及主要叶面病害,如灰霉病、黑斑病、褐斑病、黑胫病、菌核病和锈病等。使用剂量通常为200g(a.i.)/hm2,在此剂量下,活性优于或等于常规杀菌剂如氟环唑、戊唑醇、嘧菌环胺等。
灭菌唑制剂FS如25%、30%等。
灭菌唑的作用机理与特点是甾醇生物合成中C-14脱甲基化酶抑制剂,主要用作种子处理剂。
这篇文章将探讨1,2,4-三氮唑钠在化学合成领域的具体应用。
简述:1,2,4-三氮唑钠,英文名称:1,2,4-Triazolylsodium,CAS:41253-21-8,分子式:C2H2N3Na,外观与性状:白色固体。1,2,4-三唑钠盐是一种以唑为基础的抗真菌剂,可用于抑制未经调理的松木上的霉菌。
1. 合成硅氟唑
硅氟唑是含氟、含硅三唑类杀菌剂,主要是通过阻碍真菌细胞膜中麦角甾醇 的形成,而发挥药效。硅氟唑与其他三唑类杀菌剂相比,内吸性更强,杀菌谱更 广,低毒,用量少,在植物体内有良好的渗透作用,兼具灭菌与防护作用。
首先用格氏试剂法和气相光氯化法制备氯甲基三甲基硅烷,然后氯甲基三甲基硅烷与镁反应生成格氏试剂(三甲基硅基)甲基氯化镁(中间体1);2-氯-4ˊ-氟苯乙酮与三唑钠发生亲核取代生成2-(4-氟苯基)- (1H-1,2,4-三唑 -1-基)-2-酮(中间体2),其与中间体1在乙醚溴化镁催化剂下发生加成反应,最后 经饱和氯化铵水溶液水解得到目标产物硅氟唑。合成路线如下:
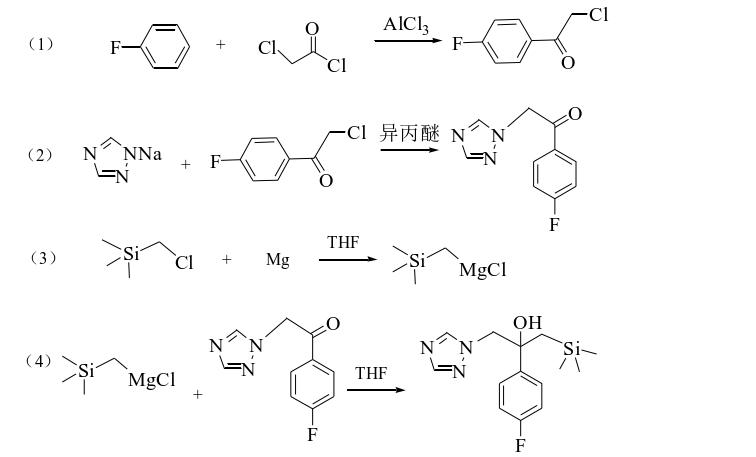
其中,三唑钠主要参与2-(4-氟苯基)- (1H-1,2,4-三唑 -1-基)-2-酮(中间体2)的合成,具体步骤如下:在氮气保护下,向装有机械搅拌的250ml干燥的四口瓶中加入5.2g(0.03mol)2-氯 -4ˊ-氟苯乙酮,80ml异丙醚,搅拌下使2-氯-4ˊ-氟苯乙酮溶解。在室温下加入3.8g三 氮唑钠(0.042mol)及0.48g(6mmol)聚乙二醇800。升温至68℃(回流)反应,反应过程中薄层色谱监控反应,直至监控无2-氯-4ˊ-氟苯乙酮原料点,降温至室温,停止反应。减压抽滤,将固体干燥后用乙酸乙酯溶解,减压过滤,除去固体后,将滤液旋干,即得目标产物2-(4-氟苯基)-1-(1H-1,2,4-三唑-1-基)-2-酮。
2. 合成噁醚唑
噁醚唑,又名苯醚甲环唑,为苯醚基环唑类新型杀虫杀螨剂,具有内吸性,是甾醇脱甲基化抑制剂,其作用位点为昆虫细胞内线粒体,不同于目前常用的杀虫剂。该产品具有杀虫谱广、活性高、持效期长、对有益生物安全和对环境友好等特点,具有触杀 胃毒作用。
苯醚酮为起始原料,经缩酮环化反应、溴代反应、亲核取代反应和粗品精制制得噁醚唑纯品。实验表明,当苯醚酮与1,2-丙二醇的摩尔比为1∶1.35,缩酮与溴的摩尔比为 1∶1.30,溴代缩酮与1,2,4-三唑钠的摩尔比为1∶1.25,缩合温度为140℃时,噁醚唑总收率可以高达83%以上,产品纯度达到98%。合成路线如下:
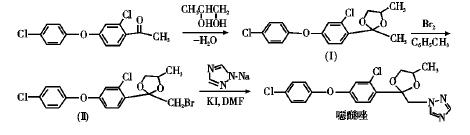
其中,三唑钠主要参与噁醚唑(±)-1-((2-(2-氯-4-(4-氯苯氧基)苯基)-4-甲基-1,3-二噁烷-2-基)甲基)-1H-1,2,4-三唑的合成,具体步骤如下:中间体II粗品(含量93.5%)约96.8 g用以N,N-二甲基 甲酰胺(DMF)和正戊烷的混合溶剂550 mL溶解,加入3.5 g KI 和一定量的1,2,4-三唑钠,搅拌,升温回流8 h。然后将正戊烷蒸出,再升温反应8 h左右,HPLC检测反应进度,反应液中噁醚唑含量达到78%以上即可停止反应,蒸出DMF,得噁醚唑粗品为粘稠液体。
参考文献:
[1]刘英浪. 硅氟唑的合成研究[D]. 河北工业大学, 2013.
[2]李敬,柴凤兰,董玉涛. 杀菌剂噁醚唑合成工艺研究 [J]. 广州化工, 2011, 39 (21): 99-101.
引言:
2-(4-碘苯基)-3-(4-硝基苯基)-5-苯基氯化四唑在细胞生物学和化学分析领域中的独特性质,使得它成为研究细胞代谢活性及染料化学反应的重要工具之一。
简介:碘硝基四氯化物(Iodonitrotetrazolium chloride,INT),即2-(4-碘苯基)-3-(4-硝基苯基)-5-苯基-2H-氯化四唑,是一种广泛用于科学研究的四唑盐。这种化合物呈淡黄色,可溶于水,还原后可形成红色甲瓒染料。INT因其特有的性质而被广泛应用于多种细胞生物学测定中,尤其是那些用于测量细胞代谢活性的实验中,成为一种极具价值的工具。

应用:
1. 测定土壤脱氢酶活性
W. von Mersi描述了使用2(p-碘苯基)-3-(p-硝基苯)-5-苯基氯化四氮唑(iodonitrotetrazolium chloride, INT)作为底物,快速、精确[100 μg氯化碘-formazan ml-1测定混合物],且易于重复测定潜在土壤脱氢酶活性的条件。经N,N-二甲基甲酰胺和乙醇萃取后,采用分光光度法(464 nm)测定还原型碘硝基四唑甲氮(INTF)含量。用这种方法所形成的有色络合物具有很高的稳定性。研究了pH、缓冲液浓度、温度、底物浓度、土壤重量和反应时间对脱氢酶活性的影响。基质水解速率与土壤重量成正比;在40℃下使用1 M TRIS缓冲液(pH 7.0)实现最佳INT降低。通过比较高压灭菌和未无菌土壤样品的测定,可以确定生物和非生物基质的还原。不同的研究证实了胞内酶与微生物生物量高度相关,并表明该活性适合作为微生物生物量的间接参数,进行测量。
Thalmann (1968) 描述的三苯基四氮唑氯化物 (TTC) 法和 Spothelfer-Maga a 和 Thalmann (1992) 描述的碘硝基四氮唑氯化物 (INT) 法用于测量土壤脱氢酶活性,现已进行改进以克服一些方法上的缺陷。建议测量波长为:溶解在丙酮中的三苯基甲臜的最大吸收值为 485 nm,溶解在四氢呋喃中的碘硝基四氮唑甲臜 (INTF) 的最大吸收值为 491 nm,溶解在 N,N-二甲基甲酰胺中的 INTF 的最大吸收值为 455 nm。用丙酮萃取三苯基甲臜两次毒性较小,并且经证实其效率至少与用 90% 丙酮和 10% 四氯化碳混合物萃取(Thalmann 1968 方法)一样高。四氢呋喃和二甲基甲酰胺从土壤中提取 INTF 的效果一样好,但前者的毒性较小。厌氧培养导致三苯甲臜和 INTF 的形成量增加,标准误差也减小。TTC 和 INT 还原均表现出较高的重现性,并且能够很好地区分六种土壤的微生物活性。由于多种原因(更容易根据不同土壤类型确定底物剂量、还原效果更好、培养时间更短),INT 还原似乎是一种比 TTC 还原更适合测量土壤微生物活性的方法。
2. 测定琥珀酸脱氢酶活性
Munujos等人描述了一种测定琥珀酸脱氢酶活性的分光光度测定方法,其中碘硝基四唑氯化物用作最终电子受体。通过测量四唑盐还原引起的甲臜的形成来确定酶活性。该测定连续、快速、简单且灵敏,可用于测定组织匀浆中的酶活性或作为细胞分馏过程中线粒体部分的标记。
参考:
[1]Von Mersi W, Schinner F. An improved and accurate method for determining the dehydrogenase activity of soils with iodonitrotetrazolium chloride[J]. Biology and fertility of soils, 1991, 11: 216-220.
[2]Friedel J K, M?lter K, Fischer W R. Comparison and improvement of methods for determining soil dehydrogenase activity by using triphenyltetrazolium chloride and iodonitrotetrazolium chloride[J]. Biology and fertility of soils, 1994, 18: 291-296.
[3]Munujos P, Collcanti J, Gonzalezsastre F, et al. Assay of succinate dehydrogenase activity by a colorimetric-continuous method using iodonitrotetrazolium chloride as electron acceptor[J]. Analytical biochemistry, 1993, 212(2): 506-509.
[4]https://pubchem.ncbi.nlm.nih.gov/compound/
氟氯西林钠,是一种β-内酰胺类抗生素,化学式为C19H16ClFN3NaO5S,分子量为475.854,性状为白色或类白色结晶性粉末,有引湿性。该物质在水中极易溶,在甲醇中易溶,在乙醇中溶解。有关该物质的部分物理数据包括:熔点176-178℃,沸点677.3oC,闪点363.4oC,蒸汽压:2.86E-19mmHg at 25°C.

氟氯西林钠最早由英国Beecham公司研发,为第四代半合成的异恶唑青霉素,通过侧链改变使之具有一个受保护的中央内酰胺环,从而具有抗青霉素酶的作用,并对胃酸稳定,能口服,具有更广的抗菌谱。它主要通过抑制细菌细胞壁黏肽的生物合成而起到强大的杀菌作用,尤其是对青霉素耐药的金黄色葡萄球菌等有很强的灭菌活性,临床上主要用于治疗敏感的革兰阳性菌引起的病症[1]。其主要制剂包括注射用氟氯西林钠和氟氯西林钠胶囊[1].
取氟氯西林钠约2mg,加水0.05mL和硫酸-甲醛试液(取甲酸溶液2mL,加硫酸100mL,混匀)2mL,混匀,溶液显黄绿色,在水浴中加热1分钟,溶液变黄色.
氟氯西林钠有抗耐葡萄球菌所产生的β-内酰胺酶能力,肌注同量的氟氯西林钠与氯唑西林,血药浓度明显较高,血中有效浓度维持时间较长.
临床试验表明,氟氯西林钠主要用于治疗耐青霉素金黄色葡萄球菌的严重感染,以及呼吸道感染(如急性咽炎、化脓性扁桃体炎)、治疗感冒继发细菌感染、急慢性气管炎、支气管炎、肺炎、肺脓肿、脓胸、骨髓炎、化脓性关节炎、急慢性中耳炎、鼻副窦炎、牙周炎、疖、痈、丹毒、蜂窝组织炎、破伤风、甲沟炎、创面及伤口感染、烧伤感染、导尿后引起的尿道炎、前列腺炎、淋病、心内膜炎、革兰阳性菌尤是金黄色葡萄球菌引起的败血症.
1、氟氯西林钠不能和粘菌素甲烷磺酸钠、庆大霉素、卡那霉素和多粘菌素B配伍使用.
2、丙磺舒可减低本药的肾脏排泄.
3、氟氯西林钠不能与氨基酸溶液、脂肪乳和血液混合.
氟氯西林钠的合成方法主要包括以下步骤:(I)在三乙胺和亚磷酸三乙酯的作用下,3-(2-氯-6-氟苯基)-5-甲基异恶唑-4-甲酸与二硫化二苯并噻唑在二氯甲烷中发生缩合反应,得到活性酯反应液;(II)向步骤(I)得到的活性酯反应液中加入水和6-氨基青霉烷酸,然后再滴加三乙胺进行酰胺化反应,反应结束后经过后处理得到所述的氟氯西林钠。该合成方法避免了酰氯中间体的使用,同时中间体不需要提纯,直接通过一锅法进行后续步骤,操作简便,并且产物的收率和纯度高,便于工业化[2].
[1]周改平.氟氯西林钠的合成工艺[J].山西医药杂志:2013, 42(12):2.DOI:CNKI:SUN:SXYY.0.2013-12-067.
[2]陈亮,厉昆,赵胜贤,等.一种氟氯西林钠的合成方法.CN201811404988.2.
二苯基N,N'-二异丙基亚磷酰胺,中文别名二异丙基胺基亚磷酸二苄酯,二苄基二异丙基胺基磷,其分子式为C20H28NO2P,分子量为345.42。关于该化合物,二苯基N,N'-二异丙基亚磷酰胺常表现为白色至淡黄色粉末状固体,有关的物性数据包括:沸点130 ℃ at 0.55mmHg(lit.),密度1.028 g/mL at 25℃(lit.),折射率n20/D 1.535(lit.),闪点158 °F。

医药化工领域报道了一种抗革兰氏阳性菌的化合物的制备方法,具体涉及一种磷酸特地唑胺的制备方法。本发明的制备方法,每一步的中间体及最终产品均具有高纯度。另外,采用二苯基N,N'-二异丙基亚磷酰胺作为磷酰化试剂,还可避免产生二聚化产物,使得本发明的制备方法具有更高收率。本发明的制备方法路线较短,反应条件温和,还避免使用有毒,有刺激性和强腐蚀性试剂,绿色环保。同时避免使用超低温反应,操作简便易制备,生产效率高。因此,本发明的制备方法特别适应工业化生产[1].
还有文献公开了一种含有光敏单元的核酸及其制备方法,包括制备得到2-硝基苯甲基(4,4'-二甲氧基三苯基)醚5-(2-O-氰乙基-N,N-二异丙基亚磷酰胺酯)的步骤。其中亚磷酰胺酯试剂(如二苯基N,N'-二异丙基亚磷酰胺)可用于制备含有光敏单元的核酸,实验表明其在制备时具有很高的反应活性,良好的底物适应性,足够的化学稳定性以及良好的光响应性,为核酸的官能化提供一种更加简洁高效的选择,简化了制备含有光敏单元的核酸的方法,提高了光降解的敏感性,降低了其制备成本,扩大了制备光敏单元的核酸的市场应用[2].
研究表明,二苯基N,N'-二异丙基亚磷酰胺可用于合成一种纳米化钛表面负载Si,Cu-TiO2抗菌型生物活性膜层。以钛板为基材,先用砂纸对试样表面进行打磨抛光,然后进行超声清洗。然后进行酸洗,酸洗结束后,用去离子水清洗室温干燥,接着高能喷丸处理,再微弧氧化处理,在电解液配制上以去离子水为为溶剂,以醋酸钙,甘油磷酸钠,硅酸钠和醋酸铜为溶质。微弧氧化处理结束后,在将调节剂涂抹在医用钛板上,调节液选择了叔丁基二甲基羟乙氧基硅烷,二(乙酰丙酮基)钛酸二异丙酯,二苯基N,N'-二异丙基亚磷酰胺,N,N-二甲基乙醇胺和2,3-环氧丙醇的混合物最后再用钴60辐照灭菌即可。最终制得的钛板自身会有很强的抗菌性,不易受到细菌感染,提高种植手术的成功率,同时还具有很强的耐磨性[3].
[1]朱益忠,张喜全,刘飞,等.一种抗革兰氏阳性菌的化合物的制备方法:CN201410546241.6[P].CN201410546241.6.
[2]刘冬生.含有光敏单元的核酸及其制备方法.CN201810363793.1.
[3]邓振南,王黎荔,刘崇星,等.一种纳米化钛表面负载Si,Cu-TiO2抗菌型生物活性膜层的制备方法.CN201810672733.8.
青霉素酶制备培养基是一种专门用于培养高产青霉素酶蜡状芽孢杆菌的培养基。它主要用于选择培养高产青霉素酶蜡状芽孢杆菌和进行青霉素酶的发酵制备。
制备培养基的方法是取31.9g本品,另取50g甘油,加热溶解于1000ml蒸馏水中,分装于500ml锥形瓶内,每瓶80ml,经过115℃高压灭菌30分钟后备用。

青霉素酶是一种能水解β-内酰胺类抗生素的酶,它采用基因重组技术构建了高效表达菌株,通过色谱层析技术获得重组β-内酰胺酶(TEM1)。该酶具有纯度高、活性高、专一性强等特点,可以有效地分解β-内酰胺类抗生素,包括青霉素G、氨基青霉素类、羧基青霉素类和脲基青霉素类。
青霉素酶是一种作用于青霉素的β-内酰胺环的酶,能使青霉素失去抗菌活性。许多青霉素耐药细菌能产生这种酶,如金黄色葡萄球菌和蜡状芽孢杆菌等。
青霉素酶是一种医疗用酶,可用于治疗一般青霉素的过敏反应,并可作为过敏性休克或严重青霉素过敏反应症患者的辅助治疗。蜡状芽孢杆菌可作为此酶的生产菌种。
β-内酰胺酶是一类能水解β-内酰胺类抗生素的水解酶,目前已发现200多种。根据β-内酰胺酶的结构和水解产物的不同,被分为A、B、C、D四类,其中青霉素酶属于A类。
青霉素酶的主要特点是可以水解青霉素、头孢菌素、肟类β-内酰胺、氯唑西林、羧苄西林和碳青霉烯类等抗生素,其活性可被克拉维酸所抑制。
目前青霉素酶主要应用于抗生素药物的无菌检验,也有用于青霉素酶生物传感器以及以青霉素酶为标记的ELISA的报道,但都属于试剂级产品,尚无工业级产品及应用的报道。
本研究的主要成果包括:
1. 通过紫外诱变和指示菌生长谱法筛选,得到一株高产青霉素酶的菌株,命名为kd-02,使发酵单位提高了291.4%。
2. 经过发酵工艺再次优化,发酵单位平均为7,500,000U/mL,最高达7,588,000U/mL。
3. 研究了无菌酶制剂的制备工艺,发酵酶液经过0.22μm膜精滤达到了无菌要求,其过滤收率为92.3%。
4. 研究了酶制剂的稳定性,25℃以下保存1个月,其酶活稳定在92%以上。
5. 研究了上述无菌酶制剂在青霉素菌渣无害化处理中的应用,可以完全去除其中的青霉素。
[1] Sorption of tylosin onto swine manure[J]. Angela C. Kolz, Say Kee Ong, T.B. Moorman. Chemosphere. 2005(2)
[2] Emerging chemicals of concern: Pharmaceuticals and personal care products (PPCPs) in Asia, with particular reference to Southern China[J]. Bruce J. Richardson, Paul K.S. Lam, Michael Martin. Marine Pollution Bulletin. 2005(9)
[3] Thermodynamically controlled synthesis of β-lactam antibiotics. Equilibrium concentrations and side-chain properties[J]. Enzyme and Microbial Technology. 1999(8)
[4] Methicillin-resistant Staphylococus aureus and vancomycin-resistant enterococci: therapeutic realities and possibilities[J]. M Michel, L Gutmann. The Lancet. 1997(9069)
[5] 刘慧娟. 青霉素酶的制备及研究应用[D]. 河北科技大学, 2009.
沙氏琼脂培养基(SDA)是一种常用于分离培养黑曲霉和白念珠菌的培养基。此外,它还可以用于一次性使用卫生用品真菌菌落总数检测。沙氏琼脂培养基的配方中,蛋白胨提供碳源和氮源,葡萄糖提供能源,琼脂是培养基的凝固剂,氯霉素则可以抑制细菌的生长。

1、将65.1g的沙氏琼脂培养基加入1L的蒸馏水或去离子水中,搅拌加热至完全溶解,然后分装至三角瓶中。将三角瓶在115℃高压条件下灭菌15分钟。
2、取样液接种于平皿中,每个平皿中加入1mL的样品,然后加入约15mL已溶化并保温至45℃左右的沙氏琼脂培养基,摇匀后凝固。将平皿置于25±2℃下培养7天。
3、在培养7天后,观察平板上的菌落数,并计算平均值。
白念珠菌是一种条件致病菌,平时存在于人体的皮肤、粘膜、消化道及其他脏器中。当机体抵抗力降低时,白念珠菌会繁殖并引发疾病。
侵袭性真菌感染的发病率不断上升,其中念珠菌感染占主要比例,尤其是白念珠菌感染。然而,由于抗真菌药物的预防性使用,非白念珠菌尤其是对氟康唑天然耐药的克柔念珠菌感染也呈上升趋势。
随着白念珠菌和克柔念珠菌对唑类抗真菌药物耐药现象的加剧,有必要深入研究其耐药机制。
本研究旨在了解白念珠菌和克柔念珠菌对常用抗真菌药物的敏感性,并对白念珠菌耐药菌株和敏感菌株的ERG3基因进行扩增、测序和生物信息学分析,以明确白念珠菌ERG3基因突变与抗真菌药物耐药之间的关系。
研究方法包括收集患者无菌体液标本,使用沙氏琼脂培养基、玉米吐温80琼脂培养基、CHROMagar念珠菌显色培养基和API 20C AUX鉴定系统进行念珠菌的分离和鉴定。从中选取35株白念珠菌和20株克柔念珠菌进行体外药物敏感性试验,并对白念珠菌ERG3基因进行PCR扩增、测序,最后应用BLAST软件进行比对分析。
研究结果显示,在163株检出的真菌中,白念珠菌占54.61%,非白念珠菌占42.94%,霉菌占2.45%。其中,非白念珠菌中克柔念珠菌的比例最高,为16.57%。
通过Logistic多因素回归分析,研究还发现性别、病程、体温、预防性使用抗真菌药物、免疫抑制剂、有创伤的检查和治疗、患者预后和地域因素是住院患者无菌体液标本中侵袭性真菌感染的独立危险因素。
[1] Candida bloodstream infections: comparison of species distribution and resistance to echinocandin and azole antifungal agents in Intensive Care Unit (ICU) and non-ICU settings in the SENTRY Antimicrobial Surveillance Program (2008-2009) [J]. Michael A. Pfaller, Shawn A. Messer, Gary J. Moet, Ronald N. Jones, Mariana Castanheira. International Journal of Antimicrobial Agents. 2011(1).
[2] Epidemiology and treatment approaches in management of invasive fungal infections [J]. James Ito, Kriengkauykiat, Sanjeet Dadwal, Kriengkauykiat. Clinical Epidemiology. 2011(Issu).
[3] Trends in the postmortem epidemiology of invasive fungal infections at a university hospital [J]. T. Lehrnbecher, C. Frank, K. Engels, S. Kriener, A. H. Groll, D. Schwabe. Journal of Infection. 2010(3).
[4] Antifungal drug resistance of oral fungi [J]. Masakazu Niimi, Norman A. Firth, Richard D. Cannon. Odontology. 2010(1).
[5] 乔祖莎. 念珠菌ERG3基因突变对抗真菌药物耐药的作用[D]. 山西医科大学, 2012.
引言:
合成Vindoline是一项具有挑战性和重要意义的化学研究课题,其合成方法和技术一直备受关注。通过不断的实验和探索,科学家们逐渐揭示了Vindoline的合成途径,为其工业化生产和药物研发提供了重要的参考和指导。
1. Vindoline 合成简介
长春碱 和长春新碱是一类双吲哚生物碱中最广泛认可的成员,因为它们在临床上用作抗肿瘤药物。至今长春碱和长春新碱仍主要从长春花植物中提取,长春花是该生物碱的唯一来源植物。由于这两种生物碱在植物体内含量很低,分别为十万分之几和百万分之几,并无法用化学合成的方法生产,使市场价格非常高。尽管其在临床上有剂量依赖的神经毒副作用,使其应用受到一定限制。但由于其广泛的临床和其它应用及改造潜力,药物化学家们仍在努力试图用化学方法合成它们,或进行结构改造以寻求高效低毒的类似物。生物学家也很早便开始研究用植物组织细胞培养的方法来生产这些生物碱。
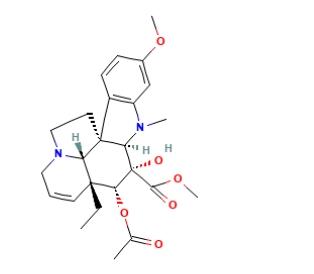
自60年代国外就开始研究长春花组织培养生产吲哚生物碱,从70年代末开始,用此来生产长春花生物碱的研究成为生物工程领域的一个热点,但至目前为止,仍未能从悬浮培养细胞中获得这两种抗癌生物碱,仅可生产阿玛碱、蛇根碱和长春质碱,也未能实现工业化生产。由于文多灵是目前半合成长春碱或长春新碱的重要前体物质,也就是说可以用长春质碱和文多灵在体外人工半合成长春碱。所以只要用植物细胞大规模培养生产出长春质碱和文多灵这两种原料,就可以半合成长春碱。Vindoline的结构如下图所示:
2. Vindoline 合成方法
可以通过两种主要方法合成:A. 全化学合成:该方法完全在实验室中从现成的起始材料构建vindoline;B. 生物合成:该方法利用基因工程微生物从较简单的前体生产vindoline。以下是每种方法的细分:
2.1 全化学合成
(1)环加成反应
这些反应通过连接两个分子形成新的环。Boger合成是一个著名的例子,它利用串联[4+2]/[3+2]环加成级联在vindoline的核心结构中同时产生三个环。
(2)立体选择反应
Vindoline有多个立体中心(具有特定空间排列的原子)。化学家在合成过程中采用各种技术来确保这些中心的正确三维排列。对映选择性催化和手性起始材料是用于此目的的一些策略。
(3)官能团操纵
在整个合成过程中,化学家引入各种官能团(如羰基、醚),并通过还原、消除、保护/去保护等反应对其进行有策略的操纵,以获得最终的vindoline结构。
2.2 生物合成:
(1)代谢工程
科学家将Catharanthus roseus(马达加斯加长春花)的vindoline生物合成途径的基因引入酵母。这使酵母具有将较简单的前体如他伯松碱转化为vindoline的酶能力。
与复杂的化学合成相比,生物合成为vindoline生产提供了一种潜在的更可持续和可扩展的方法。
3. Vindoline 的酶法合成
3.1 vindoline合成的酶促途径解释
Vindoline生物合成是在马达加斯加长春花(Catharanthus roseus)中发现的一个复杂的自然过程。它涉及从萜类中间体焦磷酸香叶基开始的多个酶促步骤。主要酶包括:
香叶醇合成酶(GES)
香叶醇8-羟化酶
8-羟基香叶醇氧化还原酶(GOR)
环烯醚萜合成酶
Tabersonine 3-加氧酶
Tabersonine 3-还原酶
3-hydroxy-16-methoxy-2 3-dihydrotabersonine-N-methyltransferase (NMT)
Desacetoxyvindoline-4-hydroxylase (D4H)
Deacetylvindoline-4-O-acetyltransferase (DAT)
这些酶共同作用,通过一系列羟基化、氧化、环化、甲基化和乙酰化反应,依次将香叶基焦磷酸转化为vindoline。
3.2 生物催化剂及其在这一过程中的作用:
在这种情况下,生物催化剂是指上面提到的酶。每种酶都像催化剂一样,是一种特殊的蛋白质,可以加速特定的化学反应而不被自身消耗。它们在vindoline合成中起着至关重要的作用:
(1)选择性
酶是高度选择性的,这意味着它们只针对特定的底物并促进所需的反应。这最大限度地减少了不必要的副反应,并允许vindoline的有效形成。与传统的化学合成相比,酶可以在温和的条件下(温度、压力、pH值)工作。这减少了能源消耗,使过程更加环保。
(2)立体控制
许多酶可以控制产物分子的立体化学。Vindoline具有具有多个手性中心的特定3D结构。酶确保这些中心的正确排列,对最终产品的生物活性至关重要。
4. 具体合成实例
4.1 方法一
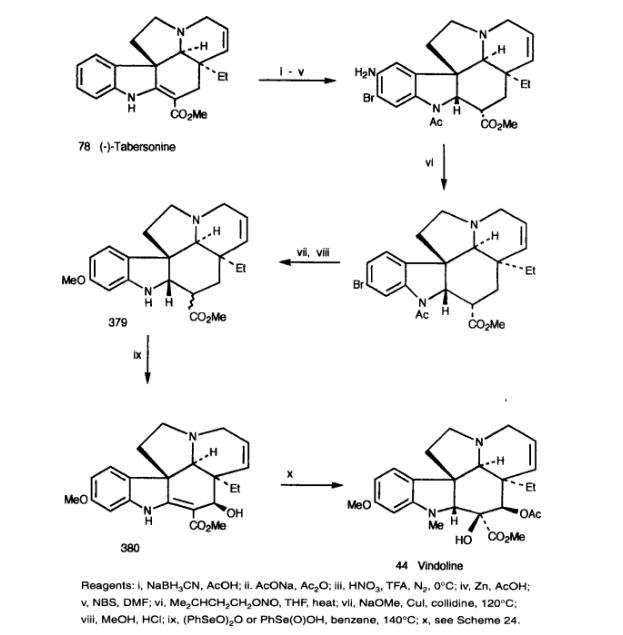
Danieli等人开发了一种将更丰富的tabersonine转化为11-甲氧基tabersonine,然后转化为vindoline的方法。由于无法清洁有效地实现对tabersonine和2,16-二氢-drotabersonine的亲电取代,因此选择N-乙酰基-2,16-二氢tabersonine作为底物。硝化反应得到10-硝基衍生物的高产率,然后通过标准工艺将其转化为11-甲氧基-2,16-二氢塔伯森碱(379),该衍生物作为C-16差向异构体的混合物获得。尝试用苯亚硒酸酐脱氢到11-甲氧基塔伯森碱偶然伴随着所需的C-17 β-羟基的引入。将产物380通过前面描述的合成长春多氨酸的方法转化为Vindoline。
4.2 方法二
4.2.1 长春花细胞系的培养
(1)长春花愈伤组织的获得
将长春花的幼茎作为外植体,用70%的乙醇表面杀菌30-50秒后投放到5-15%次氯酸钠溶液中二次杀菌10-20分钟,然后用无菌水冲洗三次,取出后在超净工作台中将幼茎切成2毫米长的茎断,接种到脱分化培养基中,在2000lx,12-16小时照光,25度左右室温下培养3-4周,即可获得白色愈伤组织。再将愈伤组织放在步骤2)所述继代驯化培养基中继代培养3次以上,同时筛选得到生长速度快,质地松软,生长量稳定的细胞系。
(2)脱分化培养基的配制
在MS培养基中,添加吲哚乙酸1.0mg/L,细胞分裂素6-BA1.0mg/L,20g/L蔗糖,然后用酸或碱将pH值调整到5.8,再添加6.5g/L琼脂,在115℃,0.1MPa压力下消毒15分钟后,经冷却制成斜面脱分化培养基。
(3)长春花愈伤组织块的继代驯化培养
将以上步骤诱导形成的愈伤组织细胞系接种在下述继代培养基中进行3次以上的继代培养,培养条件为20℃室温,避光条件,培养周期为3周。在继代培养过程中,筛选出结构疏松以及生长速度较快的愈伤组织进行继代培养;
继代驯化培养基的配制:在MS培养基的基础上添加1.0mg/L的植物细胞生长素NAA,1.0mg/L的植物细胞分裂素6-BA,20g/L蔗糖、6.5g/L琼脂,经115℃,0.1MPa压力下消毒15分钟后经冷却制成平板继代驯化培养基。
(4)长春花多倍体细胞的诱导
在MS培养基中加入10mg/L秋水仙碱(Sigma,C3915),做成液体诱导培养基,将获得的结构疏松生长速度较快的愈伤组织接种在该诱导培养基内,在20℃,避光,100rpm/分转速的摇床上振荡培养7天。收集细胞后再接种在无秋水仙碱的继代培养基中,反复驯化培养三次以上,通过生物积累量测定筛选到生长速度更快(生长速度超过5倍接种细胞量/培养周期)的多倍体细胞系;经细胞醋酸洋红或银染色后通过显微检测表明,该细胞系为混合多倍体细胞系。
(5)长春花多倍体细胞的扩增培养
将得到的长春花混合多倍体细胞系接种在扩增培养基上,在25℃,黑暗条件下进行扩增培养3周后,获得接种量10倍以上的生长旺盛的多倍体细胞;
扩增培养基的制备方法为:在改良MS培养基(按照Murashige&Skoog培养基的配方,将其中的CuSO4·5H2O和CoCl2·6H2O的添加量提高到0.1mg/L,再添加1mg/L的抗坏血酸得到的培养基)中,添加2mg/Lα-萘乙酸(NAA),2mg/L6-卞氨基嘌呤(6-BA)和30g/L蔗糖,然后用酸或碱将pH值调整到5.7,再添加4.5g/L琼脂,在115℃,0.1MPa压力下灭菌15分钟后,制成扩增固体培养基。
4.2.2 文多灵合成
将扩增培养得到的生长旺盛长春花多倍体细胞接种到合成培养基内,接种量为200g/L,在25℃,避光条件,90转/分的摇床或大型生物反应器中悬浮培养7天得到富含文多灵的细胞。收获细胞,培养液经调整后可返回反应器继续反复利用。
合成培养基:在改良MS培养基(按照Murashige&Skoog培养基的配方,将其中的CuSO4·5H2O和CoCl2·6H2O的添加量提高到0.1mg/L,再添加1mg/L的抗坏血酸得到的培养基)中,添加蔗糖30g/L,吲哚乙酸(IAA)1mg/L,KT 1mg/L,用酸或碱将pH值调整到5.8,在115℃,0.1MPa压力下消毒15分钟后冷却处理;冷却后再添加过滤除菌的组合前体(乙酰辅酶A 10μg/L、苯丙三氮唑0.5μmol/L、苯环丙胺5μmol/L、乙酸酐10μl/L、二硫苏糖醇10mg/L),用无菌的酸或碱将pH值调整到5.8,制成合成培养基;
4.2.3 文多灵的提取
将合成培养得到的富含文多灵的细胞置于提取灌中,经过高速搅拌(300转/分)破碎,加入1ml/g鲜细胞量的体积百分浓度为95%的乙醇溶液,在室温条件下萃取,然后减压(0.01MP)浓缩获得粗提物,再加入二倍体积的乙酸乙酯,用硫酸调pH值为8.0后,迅速摇荡,萃取生物碱三次,减压蒸干后获得含有文多灵的混合物。用高效液相色谱(HPLC)检测该混合物中文多灵的含量,结果表明,文多灵含量达到1mg/g细胞干重。
5. 合成 Vindoline 的应用和用途
(1)合成Vindoline的各种应用综述
合成Vindoline的主要应用在于它作为重要抗癌药物长春碱和长春新碱的前体。长春花属植物中长春花碱的天然丰度很低,化学合成也很困难,这使得合成Vindoline成为大规模生产这些救命药物的有价值的替代品。
(2)在制药和其他行业的潜在用途
除了它作为前体的既定作用之外,人们正在研究合成vindoline本身的潜在应用:
抗糖尿病特性:研究表明,vindoline可能具有抗糖尿病作用,尽管需要更多的研究来证实其作为一种治疗方法的有效性。
新药开发:合成vindoline独特的化学结构为开发具有多种治疗应用的新型药物提供了希望。通过修改vindoline分子,科学家们有可能制造出针对各种疾病的药物。
6. 结论
总的来说,合成Vindoline是一项具有挑战性和前瞻性的研究领域,其合成方法的不断完善和优化将为药物研发和生产提供重要的技术支持和指导。希望本文对Vindoline的合成方法有所启发,激发读者对这一领域的兴趣和探索。
参考:
[1]https://pubchem.ncbi.nlm.nih.gov/compound/260535
[2]https://www.sciencedirect.com/topics/chemistry/vindoline
[3]https://www.nature.com/articles/s42003-021-02617-w
[4]https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2531198/
[5]清华大学. 一种生产文多灵的方法. 2008-12-31.
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手
























