硫代乙酸钾是有机化合反应中重要的硫代试剂,具有广泛的应用。有多种方法可以制备硫代乙酸钾,其中一种方法是使用乙酐和少量氢氧化钠,在适当条件下通入H2S,然后进行快速蒸馏,将硫代乙酸和乙酸与钠盐分离,再通过精馏分离出硫代乙酸,产率为72~76%。还有其他方法,如使用乙酸和五氧化磷、乙酰氯和硫氰化钾硫、二乙酰硫水解、乙酐和硫化氢在酸性介质或在三乙胺和吡啶下反应,以及乙酰氯和硫化氢在三氯化铝存在下反应等。除了乙酐-硫化氢法外,其他方法的质量收率都不高。

为了克服现有技术的缺陷,提高硫代乙酸钾的制备效率,本研究采用了一种优化的制备方法。该方法使用双乙烯酮和多硫化钠,在无水醇类溶剂中密闭通入硫化氢气体,并加入抗氧化剂。在硫代反应完全后,通过精馏制得硫代乙酸和无水醇类溶剂。然后,将硫代乙酸或其与无水醇类溶剂的混合物滴加到氢氧化钾的无水醇类溶剂中,搅拌并减压回收无水醇类溶剂,最终得到硫代乙酸钾的白色晶体。该方法的关键在于使用抗氧化剂并控制溶剂中的水份,要求溶剂无水。该方法的收率高达90%以上,废气污染少,生产成本低。
在此制备过程中,硫代乙酸与氢氧化钾的摩尔比优选为1∶1~2,氢氧化钾与无水醇类溶剂的体积比优选为1∶3~10。
无水醇类溶剂优选为异丙醇、异丁醇、正丁醇或异戊醇,这些溶剂便于回收,能够大幅度降低溶剂成本。
多硫化钠优选为无水硫化钠或结晶体硫化钠。
抗氧化剂可以选择二丁基羟基甲苯(BHT)、丁基羟基茴香醚(BHA)或叔丁基氢醌(TBHQ),其用量为双乙烯酮重量的0.1-1%,可以将反应收率提高到几乎定量。如果不加抗氧化剂,反应收率仅为40-50%。
氢氧化钾的无水醇类溶剂中也应加入抗氧化剂,其用量为硫代乙酸重量的0.1-1%,可以进一步提高反应收率。
滴加时的温度应控制在0~5℃,搅拌时的温度应控制在60℃以下。这些反应条件简单且容易控制。
硫代的反应程度可以使用气质联用仪进行检测,当双乙烯酮的含量低于1%时,可以结束硫代反应。
本发明的反应条件温和,操作简便;反应完全,硫化氢用量少,环境污染少,收率高(达90%以上),成本低。
硫代乙酸钾是一种有机硫化合物,化学式为CH3COS?K+。它是可溶于水的白色固体,可用于制备硫代乙酸酯及其衍生物。

硫代乙酸钾是有机化合反应中重要硫代试剂,用途十分广泛。
硫代乙酸钾和烷基化试剂(如卤代烃)反应得到硫代乙酸酯,水解得到硫醇:
CH3COSK + RX → CH3COSR + KX (X = 卤素)
CH3COSR + H2O → CH3CO2H + RSH
硫代乙酸钾的制备方法,其步骤如下:
用双乙烯酮和多硫化钠在无水醇类溶剂中密闭通入硫化氢气体、加入抗氧化剂,硫代完全后,精馏制得硫代乙酸和无水醇类溶剂;将硫代乙酸或其与无水醇类溶剂的混合物滴加到氢氧化钾的无水醇类溶剂中,滴毕,搅拌,减压回收无水醇类溶剂,析出的白色晶体为硫代乙酸钾;所述的抗氧化剂为二丁基羟基甲苯、丁基羟基茴香醚或叔丁基氢醌。
本发明的技术方案为:
用双乙烯酮和多硫化钠在无水醇类溶剂中密闭通入硫化氢气体、加入抗氧化剂,硫代完全后,精馏制得硫代乙酸和无水醇类溶剂;将硫代乙酸或其与无水醇类溶剂的混合物滴加到氢氧化钾的无水醇类溶剂中,滴毕,搅拌,减压回收无水醇类溶剂,析出的白色晶体为硫代乙酸钾。本发明反应完全,硫化氢用量少,环境污染少,收率高,成本低。
CN101045701B
硫代乙酸钾,又称硫代醋酸鉀、乙硫羟酸钾盐等,是一种白色结晶粉末,熔点在173°C至176°C之间。它主要用于引入分子结构中的硫原子以及合成抗艾滋病药物等。

硫代乙酸钾的制备方法包括以下步骤:
特征如下:
a、形成硫代乙酸钾合成料:在500L的锥形螺带真空干燥机中,加入190-240kg碳酸钾或碳酸氢钾,开启螺带搅拌,加入200kg硫代乙酸,等搅拌均匀后,以喷雾方式加入10-20kg水,然后开启真空,真空度在0.0 IMPa至0.09MPa之间,升温至40-80°C,升温20-30分钟,温度达到40-80°C后,保温反应2-5小时,准备待用;
b、溶解分离:在1000L的反应锅中,加入200-800L无水溶剂,开启搅拌,放入上述合成料,搅拌半小时,待硫代乙酸钾充分溶解后,过滤;滤渣用无水溶剂洗涤,由于碳酸钾和碳酸氢钾在此无水溶剂中几乎不溶,所以滤渣即为碳酸钾或碳酸氢钾;滤渣回到合成反应中;
c、脱色:洗涤溶剂和滤液混合,加入5kg活性炭,充分搅拌后,过滤,滤渣为废活性炭,滤液加入到结晶锅中;
d、结晶:加入反应锅中,降温至(TC以下,进行结晶,过滤得到高纯度的硫代乙酸钾;由于处理过程中没有水的进入,无水溶剂不需要处理可以直接回到溶解分离步骤。
3-氟丙基磺酰氯是一种用于制备RAS/RAF/MEK/ERK和PI3K/AKT/PTEN/MTOR通路双重抑制剂的化合物。它可以通过以下步骤制备:
将3-氟丙醇与三乙胺和甲磺酰氯在二氯甲烷中反应,然后用水稀释反应混合物,并用二氯甲烷萃取。最后,使用柱色谱法纯化产物。
将3-氟丙基甲磺酸酯与硫代乙酸钾在二甲亚砜中反应,然后用水萃取反应混合物。最后,使用减压蒸发得到产物。
将氯气通过(3-氟丙基)硫代乙酸酯的反应混合物中,然后用溶液萃取产物。最后,使用减压蒸发得到3-氟丙烷-1-磺酰氯。
3-氟丙基磺酰氯可以用于制备N-(3-(2-(6-氨基吡啶-3-基)-4,8-双吗啉基喹唑啉-6-基)-2-氟苯基)-3-氟丙烷-1-磺酰胺。该化合物可作为RAS/RAF/MEK/ERK和PI3K/AKT/PTEN/MTOR通路双重抑制剂,有效地抑制肿瘤的生长。
[1] [中国发明,中国发明授权] CN201480033247.1 作为RAS/RAF/MEK/ERK和PI3K/AKT/PTEN/MTOR通路双重抑制剂的喹唑啉和氮杂喹唑啉
二硫赤鲜醇是一种具有赤式构型的二硫苏糖醇异构体,可用作巯基化DNA的还原剂和去保护剂,同时也常用于蛋白质中二硫键的还原和阻止蛋白质分子内或分子间的半胱氨酸之间形成的二硫键。
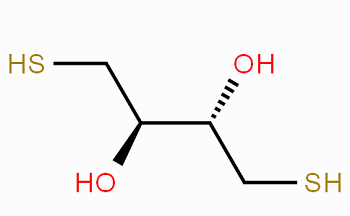
赤式构型的双羟基的构建是合成二硫赤鲜醇的难点之一。目前鲜有文献报道关于二硫赤鲜醇的合成方法。然而,CN201611235712.7成功地开发了一种制备二硫赤鲜醇的合成方法,该方法在顺式烯烃上进行顺式加成,得到赤式构型的产物。这种合成方法路线短,收率较高,具有广泛的应用价值。
该制备方法包括以下步骤:
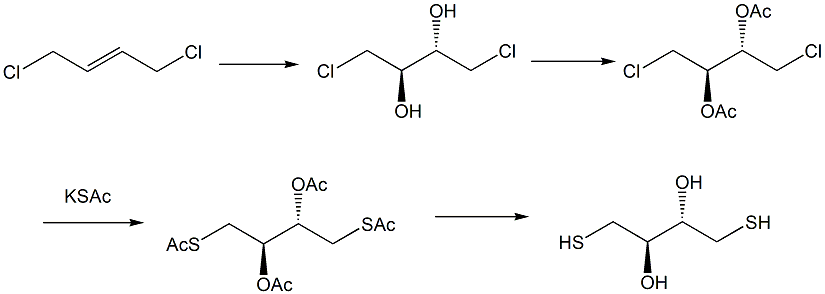
(a) 将顺-1,4-二氯-2-丁烯溶于第一反应溶剂中,在低温条件下与高锰酸钾发生顺式双羟化反应,得到式(I)1,4-二氯-2,3-二丁醇;
(b) 将步骤(a)制备的式(I)1,4-二氯-2,3-二丁醇溶于第二反应溶剂中,加入适当的有机碱,与乙酸酐发生酯化反应,得到式(II)1,4-二氯-2,3-二乙酸酯;
(c) 在第三反应溶剂中,将步骤(b)制备的式(II)1,4-二氯-2,3-二乙酸酯与硫代乙酸钾在碘化钠的催化作用下发生亲核取代反应,得到式(III)1,4-二硫代乙酰基-2,3-乙酸酯;
(d) 在第四反应溶剂中,步骤(c)制备的式(III)1,4-二硫代乙酰基-2,3-乙酸酯与酸性试剂发生水解反应,得到目标产物式(IV)二硫赤鲜醇。
反应过程如下图所示:
[1]CN201611235712.7一种二硫赤糖醇的制备方法
亚硫基二乙酸是一种羧酸类有机物,可用于医药合成中间体的制备。
1)制备一种3,4-乙撑二氧噻吩的方法:首先将亚硫基二乙酸与乙醇反应得到硫代二乙酸二乙酯,然后与对草酸二乙酯、乙醇钠反应得到3,4-二羟基噻吩-2,5-二羧酸二乙基酯二钠盐,再与盐酸反应得到2,5-二羧酸二乙酯-3,4-二羟基噻吩,接着与1,2-二溴乙烷、碳酸钾、二甲基甲酰胺反应得到二乙基2,3-二氢噻吩并[3,4-b][1,4]二恶英-5,7-二羧酸酯,最后与氢氧化钠反应得到2,5-二羧酸-3,4-乙撑二氧噻吩,最终与铜粉、喹啉反应得到3,4-乙撑二氧噻吩产品。这种制备方法具有质量好、收率高、简单易行、能耗低、成本低等优点。
2)用于一种将巯基化合物给药至哺乳动物皮肤中增白色素过度沉着区域的方法。所选的巯基化合物包括巯基乙酸、巯基丙氨酸、高巯基丙氨酸、谷胱甘肽、硫甘油、硫羟苹果酸、2-巯基丙酸、3-巯基丙酸、硫二甘醇、2-巯基乙醇、二硫苏糖醇、噻吨、硫代水杨酸、硫羟乳酸、硫代丙酸、亚硫基二乙酸、N-乙酰基-L-巯基丙氨酸、硫辛酸及其化妆品和/或药物可接受的盐。
3)制备高耐蚀三价铬黑色钝化剂的方法:该钝化剂由A溶液、B溶液和去离子水按8:3:89的容积比混合制得。其中,每升A溶液由200~250g的硝酸铬、100~150g的硝酸钴和适量的去离子水混合溶解制得;每升B溶液由80~120g的柠檬酸或丙二酸、5~50g的亚硫酸氢钠、45~250g浓度为50%的巯基乙酸铵水溶液、20~200g的亚硫基二乙酸和适量的去离子水混合溶解获得。
[1] CN201410689268.0一种3,4-乙撑二氧噻吩的制备方法
[2] CN95194154.2对哺乳动物皮肤中色素过度沉着区域的增白方法
[3] CN201510175988.X高耐蚀三价铬黑色钝化剂
对戊基环己酮是一种4-烷基环己烷衍生物,属于高档液晶材料的重要组分。该化合物能够显著提高液晶温度相区的扩大范围,增强清亮点效果,降低粘度并提高响应灵敏度。对戊基环己酮主要用于合成环己基芳环类液晶和烷基环己基缩酮,其合成过程主要通过骨架镍催化4-烷基苯酚的加氢和进一步氧化来完成。


在反应釜中加入130mL(150mmol)和80mL溶剂,然后加入预先活化的骨架镍8g。按照常规的加氢反应操作,在一定的氢压力下进行搅拌反应。待吸氢反应完成后,将反应液抽出并过滤去除催化剂,然后通过减压蒸馏去除溶剂,得到产物220.5g~24.0g,收率为81%~95%。
将Cr2O3 14.5g(95mmol)加入23.6g(240mmol)浓H2SO4中,加水稀释至50mL,然后将所得溶液滴加到38.1g(220mmol)2的500mL丙酮溶液中。反应2小时后,中和至pH=7,然后过滤固体物,用甲苯(3×100mL)进行萃取,合并萃取液,经过MgSO4干燥后蒸去甲苯,最后浓缩收集91℃/100Pa~94℃/100Pa的馏分,得到产物334.2g,收率为92%。
一项专利CN201710150932.8报道了一种选择性合成顺式4-正戊基环己硫醇的制备方法。该方法利用对戊基环己酮和硼氢化钠在四氢呋喃水混合溶剂中发生还原反应得到4-正戊基环己醇,然后与甲磺酰氯在二氯乙烷溶剂中发生甲磺酰化反应,经过后处理和结晶得到含量>98%的4-正戊基环己甲磺酸酯。接着,与硫代乙酸钾在N,N-二甲基甲酰胺溶剂中发生硫乙酰化反应,最后在甲醇水溶剂中经过低温皂化反应得到顺式4-正戊基环己硫醇,其顺式产物选择性>99%。该方法具有反应时间短、操作简单和顺式产物选择性高等优点。
[1] 杨建明,吕剑,安忠维.4-正戊基环己酮的合成[J].合成化学,2003(01):49-51.
[2] CN201710150932.8一种选择性合成顺式4-正戊基环己硫醇的制备方法
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手


























