二甲四氯钠是一种苯氧乙酸类激素型除草剂,具有白色粉末的外观,易吸湿结块。化学结构为CH?(Cl)C?H?OCH?COONa,分子质量为222.6。在水中溶解度高,可制成红褐色或棕褐色液体,pH值在9~11之间。

二甲四氯钠是高效的除草剂,可防除多种杂草,提高作物产量和品质。可与其他除草剂混用,加快杀草速度。也可用于土壤处理,减少杂草对作物生长的影响。
二甲四氯钠属于低毒类化合物,但长时间或大量接触可能导致中毒症状,如神经系统损害和肝、肾损害。
[1]徐巧明,高清,董红春,等.农药二甲四氯钠的高效液相色谱分析[J].现代农药, 2004, 3(4):2.
[2]沈国生.56%二甲四氯钠可溶性粉剂防除玉米田阔叶杂草效果[J].现代化农业, 2011.
[3]王凤芝,孙福华,张晓东.二甲四氯钠粉剂防除稻田扁杆镳草药效试验[J].河北农业科学, 2005, 009(004):40-42.
二甲四氯是一种苯氧乙酸类选择性内吸传导激素型除草剂,市场上有不同剂型和含量的产品。它可以破坏双子叶植物的输导组织,对生长发育产生干扰,导致茎叶扭曲、茎基部膨大变粗或开裂。
二甲四氯对低温和用药量最为敏感,因为它是一种激素性除草剂。超量使用或在水稻4叶期之前以及拔节之后施药容易导致药害,表现为禾苗叶片失绿发黄、新叶葱管状、穗卷曲难以抽出、穗畸形等症状。
二甲四氯需要在水稻5叶期以后才能使用,也就是在5-8叶期之间。只要在这个时期内混合使用是可以的。
二甲四氯对十字花科蔬菜(如油菜、大白菜)和棉花有较大影响。此外,2,4-滴、2,4-滴丁酯、2,4-D丁酯等除草剂也对棉花十分敏感。一旦误用,药害将不可逆转,低浓度会导致棉花停止生长,高浓度则会导致苗子全部死亡。

当水稻分蘖张开时,次生根会成乳头状群聚在基部不下扎。如果在同一块田块上连续使用二甲四氯超过20天,每亩使用50-60克纯二甲四氯钠,草多的地方会进行重复喷洒。如果是大龄水稻,每亩使用40-50克,只要不进行重复喷洒,就不会出现药害。

在移栽田中,如果使用了大剂量的二甲四氯钠,会导致根系乳突状群聚、茎扭曲、提前拔节以及茎节上长出乳突状畸形根。为什么会导致提前拔节的畸形发育,而且茎节上长出根?因为我们的水稻还没有到拔节期!右侧的图片是正常发育的同期水稻。

在水稻上,使用高剂量或在沙性山脚田中使用二甲四氯二甲胺盐会产生药害,其症状与二甲四氯相似。植株扁平,分蘖散开,老叶容易脱落,老根枯死并长出新根,容易被拨起。
农药的不当使用可能会让一年的努力付诸东流。因此,在使用农药时一定要慎重。如果发现药害,首先要确定是哪种药物导致的,然后尽快与技术员联系,看是否能够及时补救。
乙二胺四乙酸四钠四水合物一般情况下可以稳定存在,性状为白色粉末。实验研究中常用的化学物质是该物质的脱水物质,及乙二胺四乙酸四钠。乙二胺四乙酸四钠盐和铁钠盐(简称EDTA四钠盐和铁钠盐)是重要的络合剂,显影定影剂漂白剂,橡胶合成活化剂,乳化剂,分析化学螯合剂等,应用范围十分广泛[1].

针对丁苯橡胶SBR1502生产中聚合用水由脱氧水改为脱氧脱盐水后,需降低皂液中乙二胺四乙酸四钠四水合物的用量,进行小瓶聚合反应试验,考察了降低乙二胺四乙酸四钠四水合物用量对聚合反应和胶乳体系稳定性的影响。研究结果表明:在其他条件不变的情况下,降低乙二胺四乙酸四钠四水合物标准用量的10%,可以提高聚合反应转化率,增强胶乳的机械稳定性,且凝胶含量少,能够应用于工业生产中,降低装置原料成本[2].
乙二胺四乙酸四钠四水合物可用于制备水基型金属防锈剂。防锈剂由如下重量份数的原料制成:防锈剂单体:由摩尔比为1:1:2的二乙醇胺硼酸单酯,2-胺基苯并噻唑和葵二酸二甲酯合成的羧酸盐作为防锈剂单体25-35份;其中二乙醇胺硼酸单酯是由摩尔比为1:2.2的二乙醇胺和硼酸反应生成;碳酸钠2-4份;葡萄糖酸钠1-3份;异丙醇4-8份;非离子型表面活性剂为AE07 0.5-1份;乙二胺四乙酸四钠四水合物0.1-0.2份;去离子水48.8-67.4份。相关发明可在钢板表面生成均匀,完整及强度高的保护膜,作为独立相存在,把金属与溶液完全隔离,降低金属的溶解速度,从而实现对金属材料的保护,防锈效果好。并且,采用复配的生产工艺还具有常温使用、节能环保等优点[3].
以乙二胺四乙酸四钠四水合物、十二烷基磺酸钠盐、碳酸钠、盐酸、甘油、聚乙二醇等物质制备得到的清洗剂可用于火灾后金属表面处理。待处理物质经浸泡处理,高压冲洗,干燥处理过程。所述处理方法操作简单,所提供的清洗剂易于制备,且对环境污染小,对火灾后的金属表面有较强的去污能力,具有较强的经济效益,易于推广[4].
文献报道了一种二甲四氯钠的增稠组合物,其主要原料为乙二胺四乙酸四钠四水合物、表面活性剂、水等。制备得到的组合物专门用于二甲四氯钠水剂,用此组合物替代常规无增稠效果的助剂,能提高药液在叶面的粘附减少雨水冲刷,刮风引起植株叶片相互碰撞和摩擦造成的药剂流失,提高使用效果[5].
[1]潘继先.乙二胺四乙酸四钠盐和铁钠盐[J].精细化工信息, 1988(11).
[2]李涛.降低EDTA-4Na用量对丁苯橡胶性能影响的研究[J].齐鲁石油化工, 2022, 50(4):274-277.
[3]吴鲲魁,杨春杰,白金峰,等.一种水基型金属防锈剂及其制备方法:CN201710294909.6[P].
[4]赵振合.乙二胺四乙酸四钠在火灾后金属表面处理的方法:CN202110291992.8[P].CN202110291992.8.
[5]王永生,韩仲强,申宝兵,等.一种用于二甲四氯钠的增稠组合物及其制备和应用.CN201410657186.8.
氯氟吡氧乙酸是一种属于吡啶氧乙酸类的除草剂,具有内吸传导作用,属于典型的激素型除草剂。它适用于防除阔叶杂草,如猪殃殃、卷茎蓼、马齿苋等,对禾本科杂草无效。

氯氟吡氧乙酸是一种吡啶氧乙酸类内吸传导型除草剂,能够被植物叶和根迅速吸收,并在植物体内传导。它作用于植物的核酸代谢,导致植物产生过量的核酸,使部分组织转变为分生组织,导致叶片、茎和根的生长畸形,出现典型的激素类除草剂的受害症状,最终导致植物死亡。
此外,氯氟吡氧乙酸对作物安全,对耐药作物失去毒性。它在土壤中容易降解,半衰期较短,不会对后茬作物造成药害。
氯氟吡氧乙酸主要用于小麦田防除大部分阔叶杂草,如猪殃殃、荠菜、田旋花、灰绿藜等。通常与其他除草剂混用,以扩大杀草谱,提高杀草效果。它也可以与双氟磺草胺、二甲四氯钠、苯磺隆等混用。在小麦三叶一心至拔节前使用,安全性较好。
(1)防除小麦田阔叶杂草:在小麦2~4叶期,杂草出齐后,冬小麦田每亩使用200g/L氯氟吡氧乙酸乳油50~62.5mL,春小麦田每亩使用200g/L氯氟吡氧乙酸乳油62.5~75mL,兑水进行茎叶喷雾施药1次。最大残留限量为0.2mg/kg。
(2)防除玉米田阔叶杂草:在玉米田杂草2~5叶期,每亩使用200g/L氯氟吡氧乙酸乳油50~70mL,兑水进行茎叶喷雾施药1次。
(3)防除水田畦畔阔叶杂草:在空心莲子等阔叶杂草出土高峰期后,每亩使用200g/L氯氟吡氧乙酸乳油50mL,兑水进行茎叶喷雾施药1次。最大残留限量为0.2mg/kg。
(4)可以与烟嘧磺隆、硝磺草酮等药剂复配混用。
了解如何用3,5,6-四氯吡啶合成毒死蜱及其重要中间体在农药研发领域具有重要意义。
简述:3,5,6-四氯吡啶是一种重要的精细化工中间体和除草剂由2,3,5,6-四氯吡啶可进一步制备各种各样生物活性高、毒性低的除草剂、杀菌剂、杀虫剂,如农药毒死蜱。主要的制备方法有:(1)吡啶液相氯化法;(2)五氯吡啶锌粉还原法;(3)吡啶气固催化氯化法;(4)以三氯乙酰氯和丙烯腈为原料的缩合闭环法。其中气固催化氯化法流程简单,收率高而最具应用前景。
应用:合成毒死蜱及其重要中间体。
毒死蜱属于高效、低残留、中等毒性类型的有机磷杀虫剂,具有熏蒸、触杀和胃毒三重作用,也具有渗透性,叶片残留时间短,但在土壤中残留时间长,主要应用于各种农作物防治棉铃虫等多种害虫,是替代高毒杀虫剂的品种之一。毒死蜱目前是全球市场容量排名前三的杀虫剂,已在全世界大多数国家和地区登记使用。
1. 合成毒死蜱

(1)3,5,6-三氯吡啶醇钠的制备
将2,3,5,6-四氯吡啶和氢氧化钠、水按计量投入2 L反应烧瓶中,回流下进行碱解反应,得到3,5,6-三氯吡啶醇钠。
(2)毒死蜱的制备
向上述烧瓶中加入催化剂,升温,滴加O,O-二乙基硫代磷酰氯进行缩合反应,滴加过程注意控制反应温度和pH。滴加反应结束后得到含有毒死蜱的油水混合物,静置分层,放出下层油层。将油相经过水洗、酸洗过 滤,脱水后得到毒死蜱原油。缩合反应的催化剂,包括酰化反应催化剂、相转移催化剂、乳化剂三类催化剂或其任意组合,如酰化反应催化剂选用的是DMAP(4-二甲氨基吡啶);相转移催化 剂有季铵盐类和叔胺类,如四丁基溴化铵(TBAB)、苄基 三乙基氯化铵(TEBA)、十六烷基三甲基溴化铵等;乳化剂选用的是表面活性剂,如季铵盐、十二烷基硫酸钠、十 二烷基苯磺酸钾、硬脂酸钠等。
2. 制备3,5,6-三氯吡啶-2-醇钠
3,5,6-三氯吡啶-2-醇钠(sodium 3,5,6-trichloropyridin-2-ol, STCP)是合成杀虫剂毒死蜱和除草剂绿草定 (triclopyr)的重要中间体。2,3,5,6-四氯吡啶是吡啶法制备STCP的中间产物。加压条件下2,3,5,6-四氯吡啶(TCP)在间歇反应器中水解可制备3,5,6-三氯吡啶-2-醇钠(STCP)。
每次实验时在150.0 g含 0.5 wt % 氢氧化钠的水溶液中加入1.0 g TCP。所有实验是在一套如下图所示的反应装置(海安石油科研仪器有限公司)上进行的。 该装置主要由一个带电磁搅拌装置、电加热自动控温装置(温度控制精度为±1 ℃)的高压釜式反应器(200 mL) 和一个冷凝取样管组成,取样管为一段Φ3 mm×300 mm的不锈钢管。取样时,先关闭阀门2,然后打开阀门1。当取 样管中的压力和反应器中的压力平衡后,关掉阀门1。 最后打开阀门2,缓慢卸压取样于锥形瓶。TCP水解制取STCP的优化工艺条件为:TCP与氢氧化钠摩尔比约为1∶4,反应温度 140 ℃左右,反应时间3 h,收率达95%以上,产品纯度可达99%以上。

3. 合成3,5,6-三氯吡啶-2-酚
3,5,6-三氯吡啶-2-酚是生产毒死蜱的农药中间体。以五氯吡啶为原料,通过锌粉还原制备出中间产物2,3,5,6-四氯吡啶,再由2,3,5,6-四氯吡啶水解,制备出农药中间体3,5,6-三氯吡啶-2-酚。水解反应的最佳反应条件为:反应温度为95~100℃、反应时间为20h,收率可达到95%。两步反应总收率可以达到90%。具体实验步骤如下:
(1)五氯吡啶的还原
在装有搅拌器、温度计、恒压滴液漏斗、球形冷凝管的500mL四颈瓶中加入30g五氯吡啶(PCP)、250mL乙腈、Zn粉(6.26~14.87g),加热到乙腈的沸点(78℃),剧烈搅拌。待PCP全溶,于一定时间(0~60min)内滴加氯化铵水溶液(12.9g氯化铵+30g水)。滴加完毕后,维持该温度,剧烈搅拌一定时间(1.0~3.0h)。反应完毕后,维持温度在60℃以上趁热过滤出未反应的锌粉。将滤液装入500mL单口烧瓶,蒸出230mL物质(乙腈和水的共沸物)。向剩余的物质中加入6.25mol/L的盐酸100mL,搅拌冷却至室温,将烧瓶中的白色固体滤出,即得2,3,5,6-四氯吡啶(TCP) 。
(2)四氯吡啶的水解
在装有搅拌器、球形冷凝管、温度计的250mL三颈瓶中加入20g 2,3,5,6-四氯吡啶、KOH水溶液(15g KOH+100mL水),升温至95~100℃,剧烈搅拌,在该条件下反应一定时间(6~24h)。反应结束后,维持温度在80℃以上趁热过滤。向滤液中滴加6mol/L的浓硫酸,直至pH值为3~4,滴加完毕后抽滤出白色固体,用80℃以上的水反复冲洗,烘干后即得所需的产品3,5,6-三氯吡啶-2-酚(TCPO)。
参考文献:
[1]唐先龙,张涛,鲁宁宁等. 毒死蜱的合成工艺研究 [J]. 安徽化工, 2022, 48 (01): 61-65.
[2]艾秋红,刘琳琪,王良芥等. 活性炭对吡啶气固催化氯化制备2,3,5,6-四氯吡啶的影响 [J]. 湘潭大学自然科学学报, 2008, (02): 35-39.
[3]刘宁,崔洪友,姚德. 2,3,5,6-四氯吡啶加压水解制备3,5,6-三氯吡啶-2-醇钠 [J]. 农药, 2008, (01): 31-33. DOI:10.16820/j.cnki.1006-0413.2008.01.010.
[4]朱伟,肖国民. 3,5,6-三氯吡啶-2-酚的合成 [J]. 精细与专用化学品, 2005, (18): 12-14.
2,3,4,5-四氟苯甲酸是一种重要的中间体,其多种合成方法的研究对于推动药物、材料等的生产和应用具有重要意义。
背景:近年来,新一代喹诺酮类抗菌药由于其广谱,高效,毒副作用小等特点,因而发展很快。2,3,4,5-四氟苯甲酸是合成新型氟喹诺酮抗菌药的重要中间体,主要用于合成喹诺酮类药物洛美沙星、氧氟沙星、芦氟沙星、司氟沙星等,另外也广泛应用于合成树脂、导电材料以及液晶材料。其需求不断走高,而该产品合成工艺复杂、技术难度大,目前仅有少数公司能够生产,国内虽有批量生产,但质量不稳定,成本较高。
合成:
合成2,3,4,5-四氟苯甲酸的工艺路线一共有8条,按采用主要原料来分,它包括:四氯苯酐法,四氟苯甲醇法,邻苯二腈法,四氟苯法,八氯萘法,N-苯基四氯邻苯二甲酰亚胺法等。
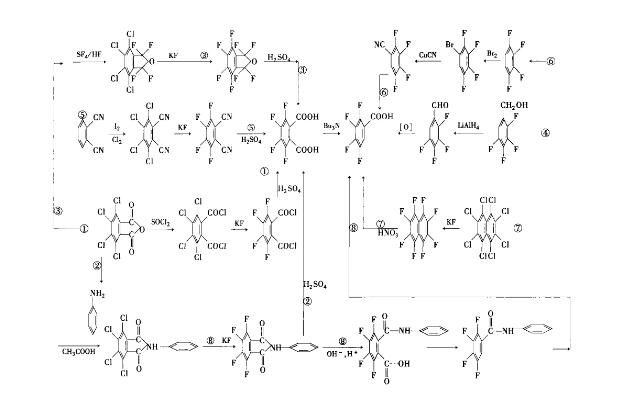
有3条起始原料都是四氯苯酐,其中路线②是适合我国国情的工业化路线。从四氯苯酐出发可分:①四氯苯酐经酰氯化得四氯邻苯二甲酰氯,然后经氟化、水解、脱羧反应得到产物; ②四氯苯酐经酰亚胺化、氟化、水解得产物;③四氯苯酐经两步氟化反应,然后水解、脱羧得产物。路线⑧是以N-苯基四氯邻苯二甲酸亚胺为起始原料的路线,原料易得、成本低,在工业上广泛采用。
1. 3,4,5,6-四氯邻苯二甲酸酐法:
3,4,5,6-四氯邻苯二甲酸酐和苯胺在常压下反应制得2,3,4,5-四氟苯甲酸。路线②步骤如下:
①缩合反应,即苯胺与3,4,5,6-四氯邻苯二甲酸酐在乙酸中、在氮气保护下化回流6h,得到产物,经过滤、水洗、烘干,得缩合反应物,熔点271℃,收率97.5%;
②氟代反应,将上得到的缩合物与氟化钾在N,N-二甲基甲酰胺(DMF)和催化剂PEG6000作用下反应10h,反应温度为150℃,原料转化率100%,产物生成率86%,收率为60%;
③开环反应,将上得到的氟化物,在氢氧化钾水溶液中反应1 h,温度控制在95℃,冷却至25℃,滴加浓盐酸析出白色固体开环化合物,经过滤、水洗、干燥得产物,熔点195℃,收率98.0%;
④脱羧反应,将上得到化合物加入到二甲基亚砜中(DMSO),在氮气保护下反应10 h,反应温度为130℃,原料转化率为92%,脱羧物生成率为82%,反应物经纯化处理得到含量为99%的产物,熔点151℃,收率61.7%;
⑤水解反应,将上得脱羧化合物加入到有氢溴酸的反应器中,在130℃反应24 h,过滤,滤饼用10%氢氧化钠洗涤后用乙酸乙酯萃取,分出有机相,水相用浓盐酸将产物全部析出,过滤、洗涤,真空干燥得产物,熔点88~88.6℃。含量99%,收率85%。
2. N-苯基四氯邻苯二甲酰亚胺法(路线⑧)
温新民等提出了在常温、常压下的合成方法研究;氟化反应采用无氮氛围下低温催化,氟盐氟化法对设备要求低, 避免了因高温不易控制而有爆炸之虑;氟反应中催化剂PEG-6000效果优于季铵盐。具体步骤为:
①氟代反应,N-苯基四氯邻苯二甲酰亚胺,在KF,PEG-6000、DMF反应条件下,控制温度为145℃,保温8 h,冷却,减压回收DMF,加入冷却水,于15~25℃搅拌反应1 h,静置、抽滤、烘干得产品,收率为96%;
②开环反应,氟化物是在碳酸氢钠、活性炭条件下反应,以后经静置、过滤、烘干得产物,熔点195℃,收率达98.0%;
③脱羧反应,从上得开环化合物在三正丁胺条件下,在130℃保温反应6 h,回收三正丁胺后,加入甲苯升温回流0.5 h,降温、抽滤、烘干得含量≥96%,收率为72%的产物;
④水解反应,将羧化物在冰乙酸、浓硫酸的水溶液中,在115℃反应6 h,降温至30℃,加水后用甲苯萃取,合并甲苯液,水洗,有机层用碳酸氢钠反萃取,水层用盐酸调pH=1-2析出白色固体,在0~5℃静置,真空干燥后得含量≥98.5%,收率为84.4%的产品。
参考文献:
[1]蔡星伟,赵玉媛,姜大伟等. 2,3,4,5-四氟苯甲酸的合成及工艺优化 [J]. 精细石油化工, 2012, 29 (03): 52-54.
[2]蔡星伟,冯亚青,齐轩等. 2,3,4,5-四氟苯甲酸的制备 [J]. 化学工业与工程, 2006, (04): 320-322.
[3]徐兆瑜. 2,3,4,5—四氟苯甲酸的合成工艺进展与展望 [J]. 化工科技市场, 2006, (07): 30-33.
本文将介绍2,4,5-三氟苯甲酸的合成方法,旨在为2,4,5-三氟苯甲酸的合成提供参考依据。
简述:克林沙星(Clinafloxacin)是一种新型氟喹诺酮类药物,其抗菌谱广,疗效好,2,4,5-三氟苯甲酸(2,4,5-TrifluorobenzoicAcid,)是合成克林沙星等氟喹诺酮类抗菌药的关键中间体,也可用于合成染料和除草剂的合成。
合成:
1.方法一:
以N-苯基四氯邻苯二甲酰亚胺为原料,通过氟代、脱氟、水解、脱羧四步反应制备2,4,5-三氟苯甲酸,含量98.6%,总收率67.7%。具体实验步骤如下:
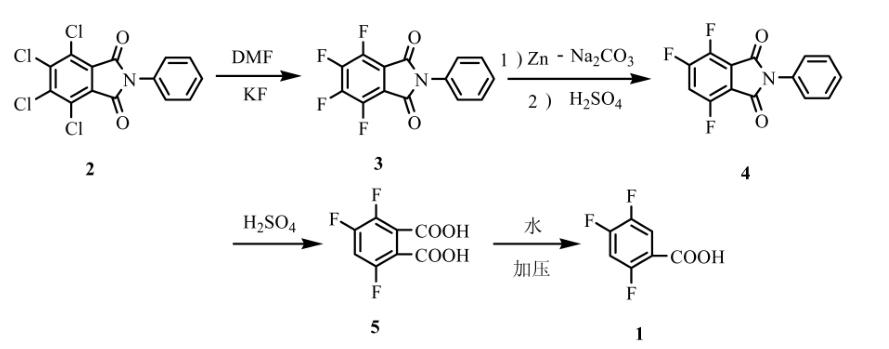
(1)N-苯基四氟邻苯二甲酰亚胺的制备(3)
将N-苯基四氯邻苯二甲酰亚胺2(含量99.35%)(72.2g,0.2mol)、氟化钾(55.8g,0.96mol)、N,N-二甲基甲酰胺(DMF)300mL投入四口反应瓶中,搅拌升温至150℃反应4h后,冷却至室温,抽滤,滤饼用DMF30mL淋洗,滤液在搅拌下缓慢加入冰水900mL,抽滤,水洗滤饼,冰乙酸重结晶,得淡黄色固体3(54.3g),熔点:204℃~206℃,收率为:88.0%,含量:96.7%(HPLC,面种归一法)。
(2)3,5,6-三氟邻苯二甲酰亚胺的制备(4)
将3(44.2g,0.15mol),水200g投入四口反应瓶中,搅拌在室温情况下滴加30%的碳酸氢钠溶液调节pH至11,控制体系pH为11,室温情况下分批加入锌粉(19.6g,0.30mol),加毕,水温升温至40℃保温反应4~6h(在保温反应过程中维持pH为11),反应完毕,降温至5℃,抽滤,滤液用硫酸调pH至3,升温至40℃保温1h后再降至室温,用4-甲基-2-戊酮150mL×2萃取,水洗有机层,无水硫酸钠干燥,减压回收溶剂至干得类白色固体4(50.0g),此固体直接用入下一步反应。
(3)3,5,6-三氟邻苯二甲酸的制备(5)
将50%硫酸溶液200g加入到上一步所得的类白色固体4(50.0g)中,搅拌升温至微回流反应8h,HPLC跟踪反应原料消失,降温,用30%的氢氧化钠溶液调pH到3~4,用4-甲基-2-戊酮150mL×2萃取,水洗有机层,减压回收溶剂至干得白色固体5(29.0g),以上两步总收率为:83.6%,含量:95.3%(HPLC,面种归一法)。
(4)2,4,5-三氟苯甲酸的制备(1)
5(46.2g,0.2mol),水300mL加入到500mL高压反应釜中,用氮气置换内的空气三次后,关闭所有的阀门,搅拌升温至140℃反应2h后,内部压力会升至10kg压力,此时反应基本完全,冷却至室温,缓慢排压后,倒出物料,用4-甲基-2-戊酮200mL×2萃取,减压回收溶剂至干,得浅白色固体,用石油醚重结晶,干燥得白色晶体1(32.4g),熔点:100.8℃~101.5℃。
2.方法二:
用2,4-二氯-5-氟苯甲酸和氯化亚砜反应得到的酰氯,经氨水酰胺化后脱水生成2,4-二氯-5-氟苄腈,再经氟代、水解制得2,4,5-三氟苯甲酸,总收率46%。具体实验步骤如下:

(1)2,4-二氯-5-氟苯甲酰氯(3)
2(93.6g,0.45mol)和氯化亚砜(200ml,2.75mol)中加入催化量DMF(0.5ml,6mmol),加热回流反应4h后常压蒸除过量氯化亚砜,加入甲苯(50ml)后减压蒸干,得黄色油状3,直接用于下步反应。
(2)2,4-二氯-5-氟苯甲酰胺(4)
0℃搅拌下向如上所得的3中滴加25%~28%氨水(360ml,5.29~5.93mol),滴毕升至室温反应12h。抽滤,滤饼用水洗后减压干燥,得白色粉末状4(80.2g,86.1%),mp157.2~158.1℃。
(3)2,4-二氯-5-氟苄腈(5)
将4(120克,0.53摩尔)和甲苯(288毫升)加热至回流,缓慢滴加三氯氧磷(30毫升,0.33摩尔),滴加完毕后继续回流反应6小时,随后用20%氢氧化钠溶液调节至pH7,进行抽滤,将滤渣丢弃。将滤液水相用甲苯(10毫升×3)进行萃取,将甲苯相合并后经减压蒸发除去溶剂,得到残渣后冷却,得到棕红色5粗品,经环己烷重结晶后得到白色固体5(90.1克,收率82.1%),熔点为44.9~45.2℃。
(4)2,4,5-三氟苄腈(6)
溴化三苯基丁基膦(4g)、环丁砜(120ml)和甲苯(50ml)加热共沸脱水,加入经喷雾干燥的氟化钾(29g,0.5mol)的甲苯溶液(50ml),加热共沸至蒸出的甲苯澄清后,加入减压干燥的5(38g,0.18mol),氮气保护下升温至150℃反应2.5h,反应过程中剧烈搅拌,GC跟踪反应至5消失后继续升温至190℃。再反应3.5h,冷却后过滤,滤渣用丙酮洗涤,滤液及洗液合并,常压除去溶剂后减压蒸干,得无色液体6粗品(22.7g),精馏收集75~77℃/3.4kPa馏分,得无色液体6(22.0,70%)。继续在80~130℃/3.4kPa蒸馏收集环丁砜和2-氯-4,5-二氟苄腈回收套用。
(5)2,4,5-三氟苯甲酸(1)
向回流状态下的75%硫酸中滴加6(20g,0.13mol),滴毕于180℃反应2h。冷至100℃以下,转至冰水(100ml)中,过滤,滤饼干燥后得白色1粗品(21.3g),用石油醚重结晶,得无色晶体1(20.7g,93.6%),mp100.3~101.1℃。
参考文献:
[1]黄生建. 2,4,5-三氟苯甲酸的合成 [J]. 浙江化工, 2013, 44 (03): 5-6+10.
[2]张明泉. 2,4,5-三氟苯甲酸合成研究[D]. 浙江大学, 2005.
齐拉西酮是一种有效的药物,被广泛应用于治疗焦虑和抑郁症等精神疾病。在本文中,我们将探讨如何合成和制备这种重要药物。
背景:以往公开的合成工艺有很多,但从起始原料与关键的反应步骤考查大致可分为以下两条制备路线。
方法一:以6-氯吲哚-2-酮为起始原料,经付克反应引入氯(或者溴)乙酰基,接着还原羰基得到5-(2-氯(或者溴)乙基)-6-氯-1,3-二氢-吲哚-2-酮然后在碱存在下与1,2-苯并异噻唑-3-哌嗪基反应得到齐拉西酮。如:EP0568619;US5206366;US5388846;US4831031 等。其反应式如下:

方法二:以2,5-二氯甲苯为起始原料,经硝化后与叔丁氧基-双 (二甲基胺基)甲烷缩合得到烯胺,接着与3-哌嗪基-1,2-苯并异噻唑反应。然后与丙二酸酯反应再脱羧,最后用亚硫酸氢钠还原关环得到齐拉西酮。如:US6111105;US5359068。其反应式如下:

方法一采用了三乙基硅烷在三氟乙酸中还原羰基,这一步反应的成本相当高,导致成本相对偏高。而方法二虽然原料的成本比较低,但其中有几步反应收率太低,特别是最后一步用亚硫酸氧钠还原关环收率只有 40%。因此,寻找一条更为方便、高效的生产途径就显得十分重要。
合成:
1. 专利CN 102250083A发明了一种齐拉西酮制备方法,其特征是,(1)向反应容器中加入水溶性的极性非质子性溶剂,开搅拌,投入5-(2-氯乙基)-6-氯-1,3-二氢-吲哚-2-(2H)-酮和3-(1-哌嗪基)-1,2-苯并异噻唑盐酸盐,再加入无机碱的水溶液,然后升温至60~70℃,保温反应3~6小时;所述5-(2-氯乙酰基)-6-氯-1,3-二氢-吲哚-2-(2H)-酮与3-(1-哌嗪基)-1,2-苯并异噻唑盐酸盐的摩尔比为1∶1~1.3;所述水溶性的极性非质子性溶剂与无机碱的水溶液的体积比为7∶3~8∶2;(2)反应完毕后,降温至30℃,加入纯化水,20℃~30℃搅拌30~60分钟,抽滤,洗涤,干燥制得齐拉西酮;所述纯化水的加入量为水溶性的极性非质子性溶剂体积的1~3倍。
该发明反应溶剂采用水溶性的极性非质子性溶剂和无机碱的水溶液的混合溶剂,既增加了反应物的溶解性,又增加了缚酸效果,缩短了反应时间 ;反应条件简洁,操作简单,易于工业化生产;后处理简单,产物收率高,杂质含量少。
2. 专利CN 101450946A发明了一种合成齐拉西酮的新方法。 该方法包括以下步骤:以 2,5 - 二氯甲苯为起始原料,经硝化后与N,N-二甲基二甲氧基甲胺缩合。所得中间体在草酸及乙二醇存在下转化成缩醛化合物。 接着跟丙二酸二乙酯反应,然后脱羧、还原、环合得到关键中间体7。 化合物7 在酸性条件下脱保护得到醛8,醛8在三乙酰氧基硼氢化钠存在下与3-哌嗪基-1,2-苯并异噻唑反应得到齐拉西酮。该发明的反应容易控制而且简洁,原料易得,操作方便。
有机溶剂为苯、甲苯、二甲苯、二氧六环、乙腈、二氯甲烷、二氯乙烷、四氯乙烷、氯仿、DMF、DMSO、N,N-二甲基乙酰胺或者N-甲基吡咯烷酮。酸性溶液为 HCl或乙酸和 HCl的混合溶液。
参考文献:
[1] 齐鲁天和惠世制药有限公司. 一种齐拉西酮制备方法:CN201110220524.8[P]. 2011-11-23.
[2] 浙江海正药业股份有限公司. 齐拉西酮的合成方法:CN200710194914.6[P]. 2009-06-10.
[3] 杭州盛美医药科技开发有限公司. 一种齐拉西酮的制备方法:CN200610125949.X[P]. 2007-07-11.
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手
























