新戊酰基乙酸甲酯又被称作4,4-二甲基-3-氧代戊酸甲酯,特戊酰基乙酸甲酯,其化学式为C8H14O3,分子量为158.19,通常情况下表现为无色至淡黄色液体,是一种密度接近于水(0.98)的具有较低水溶解度的酯类化合物。需要注意的是,由于该物质具有一定刺激性,因此在取用时需要注意防护,以免对眼睛、呼吸道和皮肤产生刺激作用。

新戊酰基乙酸甲酯的主要用途是作为一种有机合成中间体合成如草甘膦等农药、药物和染料等有机化合物。此外,该物质还可以作为溶剂和涂料添加剂在化妆品和香料行业中使用。
此外,新戊酰基乙酸甲酯还是成色剂中间体,是新型感光材料中间体,可应用于印刷制版领域,如CTP、PS版等。还可用于半导体制造,如硅晶片制造等。
新戊酰基乙酸甲酯可以通过酯化反应得到,常用的制备方法是将甲醇和3-氧代戊酸反应,反应过程中加入一定的酸催化剂,使得反应发生。
除以上提到的通用方法,也可以频那酮和过量的碳酸二甲酯为原料在二甲苯溶液中经一步反应,制得纯度99%以上的新戊酰基乙酸甲酯。对合成条件进行探讨,确定原料配比(频那酮/碳酸二甲酯)为1:5.0,反应温度为120℃,催化剂用量(甲醇钠/频那酮)为1.5:1,在优化条件下,产品的收率可(以频那酮计)达77.4%。
另外一种制备方法同样以频那酮为起始原料,辅以氢化钠和碳酸二甲酯,甲苯为溶剂,在55~60℃下,搅拌反应后冷却,在pH=4~5时制备得到新戊酰基乙酸甲酯粗品;其粗品在真空度小于10mmHg,温度为90~95℃条件下进行蒸馏可以得到收率高、产品纯度高的目标物质。其主含量达98%以上,总收率亦达96%。
[1]杨忠愚,潘伟春,孙楠,胡惟孝.感光材料中间体4,4-二甲基-戊酮-3-酸甲酯的合成[J].精细化工, 1998, 15(1):49-41.
[2]孙延晖,姚成.特戊酰乙酸甲酯的合成研究[J].化工时刊, 2001, 15(5):4.DOI:10.3969/j.issn.1002-154X.2001.05.009.
[3]曹爱春,赵松雪,郁培华.4,4-二甲基-3-氧代-戊酸甲酯的制备方法:CN201610739793.8[P].CN106397200A.
4-氨基-2-氯烟醛是一种常用的医药合成中间体。当接触到4-氨基-2-氯烟醛时,应采取相应的应急措施。如果吸入,请将患者移到新鲜空气处;如果皮肤接触,请脱去污染的衣着,用肥皂水和清水彻底冲洗皮肤,如有不适感,应就医;如果眼睛接触,请分开眼睑,用流动清水或生理盐水冲洗,并立即就医;如果误食,请立即漱口,禁止催吐,并应立即就医。

制备4-氨基-2-氯烟醛的过程可以分为以下几个步骤:
首先将4-氨基-2-甲酰吡啶溶解于二氯甲烷中,然后加入新戊酰氯溶液,在室温下反应一段时间。反应结束后,通过减压浓缩和硅胶柱色谱法纯化,得到N-(2-氯-4-吡啶基)-2,2-二甲基丙酰胺。
将2,2,6,6-四甲基哌啶和正丁基锂在四氢呋喃中反应,然后加入N-(2-氯-3-甲酰基-4-吡啶基)-2,2-二甲基丙酰胺的四氢呋喃溶液。反应结束后,通过乙酸乙酯萃取和硅胶柱色谱纯化,得到N-(2-氯-3-甲酰基-4-吡啶基)-2,2-二甲基丙酰胺。
将N-(2-氯-3-甲酰基-4-吡啶基)-2,2-二甲基丙酰胺在盐酸中反应,然后通过乙酸乙酯萃取和减压浓缩,得到4-氨基-2-氯烟醛。
[1] WO2001032658 POLYAZANAPHTHALENE COMPOUND AND MEDICINAL USE THEREOF
本文将探讨(Z)-2-(2-叔丁氧羰基氨基噻唑-4-基)-2-戊烯酸的合成方法及其在应用中的潜力。通过分析目前的合成路线和存在的问题,将提出改进方向和解决方案。
简介:化合物(Z)-2-(2-叔丁氧羰基氨基噻唑-4-基)-2-戊烯酸是合成头孢卡品酯的关键中间体。头孢卡品酯化学名为7-[2-(2-氨基-1,3-噻唑-4-基)戊-3-烯酰胺基]-3-氨基甲酰氧甲基-8-氧代-5-硫-1-氮杂 双环[4.2.0]辛-2-烯-2-甲酸2,2-二甲基丙酰氧甲基酯盐,由日本盐野义制药株式会社开发。头孢卡品酯是第4代可口服头孢类抗生素。药理研究结果表明,头孢卡品酯较现有口服的头孢品种相比抗菌活性强,剂量小的特点。因此,深入研究头孢卡品酯关键中间体(Z)-2-(2-叔丁氧羰基氨基噻唑-4-基)-2-戊烯酸化合物的合成方法,有着重要的学术意义和广阔的应用前景。
1. 合成:
1.1 目前(Z)-2-(2-叔丁氧羰基氨基噻唑-4-基)-2-戊烯酸的合成路线主要是以4-氯乙酰乙酸乙酯为原料。首先,它与硫脲反应生成环状产物,然后使用碳酸二叔丁酯保护氨基,并进行水解得到目标产物。但是4-氯乙酰乙酸乙酯与硫脲关环时活性低,导致产物收率难以提高;直接使用4-溴乙酰乙酸乙酯进行反应虽然能增加反应物的活性,但原料成本较高,并且在2缩合反应中副产物也增加。此外,在2缩合反应过程中,文献报道都是使用无机碱和有机碱进行催化,但产物收率较低,色泽不佳,纯度不高。
1.2 以4-氯乙酰乙酸乙酯为原料,首先与丙醛缩合后,由溴化钠进行氯原子交换制得溴化物,再与硫脲成环,碳酸二叔丁酯保护氨基,最后乙酯水解制得(Z)-2-(2-叔丁氧羰基氨基噻唑-4-基)-2-戊烯酸目标产物,收率达到76%,纯度达到98%。该方法简单易行,收率高,纯度好,是一条适合工业化生产的新合成方法。
2. 应用:合成头孢卡品酯。头孢卡品前体酸是合成盐酸头孢卡品酯的重要中间体,以7-氨基头孢烷酸为起始原料,经水解反应得3-脱乙酰基-7-氨基头孢烷酸,再与(Z)-2-(2-叔丁氧羰基氨基噻唑-4-基)-2-戊烯酸、二异丙胺作用得到头孢卡品前体酸。总收率70%以上,纯度98%以上。具体步骤为:

2.1 HACA的合成
在三口烧瓶中,加入甲醇100mL,纯化水100mL,7-ACA 20.4g(0.075 mol),-10℃加入11.5% (w/v)的氢氧化钠30mL,该温度下反应2h,升温至0~ 5℃,缓慢滴加3M盐酸25mL,养晶1h,抽滤,50mL 甲醇洗涤,真空干燥(<50℃),得白色固体15.4g,产率89.3%。
2.2 头孢卡品前体酸的合成
反应瓶1:加入二氯甲烷85 mL,HACA 11.5 g(0.05moL),二异丙胺8.5 mL 0~5℃搅拌至溶清得 ①,备用。
反应瓶2:加入二氯甲烷133 mL,BAPA 16.4 g(0.055 moL),<-30℃加入TEA 8 ml,甲烷磺酰氯4.5 mL,-30℃反应3 h,保持-30~-35℃加入①,-35℃反 应2h,-35℃以下加入二异丙胺(DPIA)4.2 mL,反应 1h,加入甲烷磺酸3.2 mL,反应0.5 h,-30℃缓慢滴加氯磺酰异氰酸酯(CSI)7 mL,TLC跟踪反应进度,反应完毕加入DMF48 mL,8%(w/v)氯化钠溶液160 mL,0~5℃反应2 h,分相,水相弃,收集有机相。
有机相加入丙酮200 ml,20~25℃1 h内滴加二异丙胺9 ml,20~25℃养晶3 h,降温至0~5℃,养晶 1 h。抽滤,丙酮50 ml洗涤,真空(<50℃)干燥,得淡黄色固体26.0 g,产率80.2%。
参考文献:
[1]唐磊,贾玲晓,张淑婷等.头孢卡品前体酸的合成[J].天津化工,2013,27(02):4-5.
[2]孙会,汪祝胜,潘镇浩等.(Z)-2-(2-叔丁氧羰氨基噻唑-4-基)-2-戊烯酸的合成[J].化工生产与技术,2012,19(06):25-27+8.
本文旨在探讨他达拉非中间体如何参与合成他达拉非。通过深入研究这一合成过程,有望为相关领域的发展提供新的见解和启发。
背景:他达拉非的结构由四元杂环及亚甲二氧基苯组成,经分析,他达拉非可以通过以下片段合成:D-色氨酸甲酯母核,胡椒醛母核,氯乙酰氯及甲胺。关于他达拉非的合成路线有很多文献报道,综合文献报道的方法,他达拉非的合成主要途径多是以D-色氨酸甲酯和胡椒醛为起始原料,通过皮克特-施彭格勒(Pietet-Spengler,简称P-S缩合)反应制备顺式四氢咔啉衍生物,然后再与氯酰氯和甲胺反应得到他达拉非。
(1R,3R)-1-(1,3-苯并二氧戊环-5-基)-2-(氯乙酰基)-2,3,4,9-四氢-1H-吡啶并[3,4-B]吲哚-3-羧酸甲酯(T6)是合成他达拉非的重要中间体。
合成他达拉非:
(1)

此路线是专利WO9519978报道的最早合成他达拉非的路线,以D-色氨酸甲酯和胡椒醛为起始原料,经缩合、酰化、环合等三步反应得到他达拉非。此路线存在以下缺点:①用到三氟乙酸(TFA)作为催化剂,该试剂对设备及人员的腐蚀性强、同时反应的总收率为25%,偏低。②Pietet-Spengler缩合反应的立体选择性差,反应得到顺式和反式的四氢咔啉的混合物(T4和T5),需经柱层析分离,才能得到纯度较高的顺式四氢咔啉(T4),缩合反应的收率为42%。因此,该工艺反应时间长、异构体分离困难、成本较高,不适合工业化生产。
(2)

此路线是Revell等人报道的合成路线,以D-色氨酸甲酯盐酸盐和胡椒醛为起始原料,经缩合、环合、脱保护等三步反应合成他达拉非。此路线不同之处是将Pietet-Spengler反应与酰化反应合并为一步,但是仍然需要面对立体选择性差的问题,需要使用到柱层析来分离顺反异构体,反应总收率偏低(28%),同时需要用到较为昂贵的Fmoc-Sar-Cl(芴甲氧羰基肌氨酰氯),成本较高,不适宜工业化生产。
(3)

此路线是张云龙等人报道,以D-色氨酸甲酯盐酸盐和胡椒醛为起始原料,在浓盐酸与甲醇的回流体系下,反应生成一种顺式和反式的四氢咔啉衍生物的混合物(T4和T5),经冷却沉淀出顺式体,通过过滤即可获得较为单一的目标产物(T4),T4合成步骤收率85%,该方法虽然改进了纯化方法简化了操作,但T4合成收率仍然偏低。
(4)

此路线是Orem等人报道的文献,几乎得到单一的顺式产物的新合成方法,D-色氨酸甲酯盐酸盐(T2)与胡椒醛(T3)在异丙醇单一溶剂中进行P-S缩合反应,生成的顺式四氢咔琳衍生物(T4)在异丙醇中溶解度极低,随着反应的进行从溶液中析出,该步的产率为92%,经过滤即可得到顺式产物T4。
参考文献:
[1]孙玲玲. 他达拉非的合成及其相关杂质研究[D]. 河北师范大学, 2017. DOI:10.27110/d.cnki.ghsfu.2017.000021
[2]刘文杰. 他达拉非的合成工艺研究[D]. 兰州大学, 2017.
本文旨在探讨合成(±)-3-氨甲基-5-甲基己酸的方法。通过深入研究这一合成过程,有望为相关领域的发展提供新的见解和启发。
背景:普瑞巴林(Pregabalin,PGB)是美国辉瑞 (Pfizer)公司研制的一种新型γ-氨基丁酸受体阻滞剂,已经成为治疗神经痛更高效、安全、方便的药物之一。(±)-3-氨甲基-5-甲基己酸是抗惊厥、抗癫痫药物普瑞巴林的重要中间体。
合成:
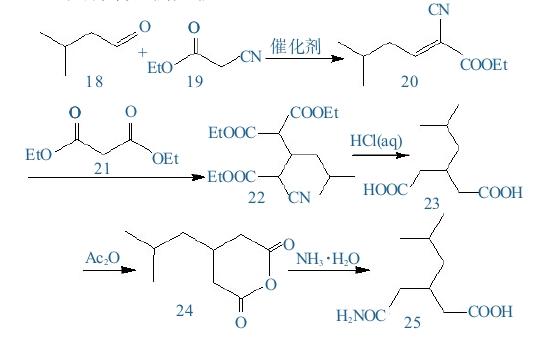
(1)3-异丁基戊二酸的合成
将22.6 g氰乙酸乙酯和50 mL正己烷加入到 250 mL反应瓶中,搅拌均匀(静置时有分层现象),再加入0.2 g(催化剂量)三乙烯二胺,搅拌均匀,控制温度在0~5℃,缓慢滴加17.2 g异戊醛,升至室温,搅拌1 h,溶液颜色加深。
用盛有正己烷的分水器加热至90℃,回流分水,减压浓缩,除去正己烷,得到36.5 g浅黄色油状物2-氰基-5-甲基-2-烯-己酸乙酯。该产品收率约为99.94%,无需后处理,可直接进行下步反应。
在上步产物中直接加入35.2 g丙二酸二乙酯和 2.02 g三乙烯二胺,加热至45~50℃,搅拌,反应10 h。反应结束后得到红棕色油状物3-异丁基 -2-氰基-4-乙氧羰基-戊二酸乙酯。
在上步反应物中直接加入200 mL、7.2 mol/L 盐酸,加热回流72 h,反应液冷却至室温,用160 mL二氯甲烷进行萃取,减压浓缩,得到35.5 g红棕色油状物3-异丁基戊二酸粗产品。该粗产品收率约为94.41%。
将提取后的残余物进行下一步反应,得到3-异丁基戊二酸,最终折算收率约为71.25%。
(2)3-(氨甲酰甲基)-5-甲基己酸的合成
称取18.8 g 3-异丁基戊二酸加入到100 mL反应瓶中,直接加入15.3 g乙酸酐,在135℃下反应 4 h,反应结束后,降温,减压浓缩,除去多余的乙酸酐和反应产生的乙酸,得到浅黄色油状物3- 异丁基戊二酸酐粗品。
在250 mL反应瓶中,加入50 mL、10%的氨水冷却至0~5℃。温度控制在0~5℃,缓慢滴加 40 mL 3-异丁基戊二酸酐的甲基叔丁基醚溶液,滴加完毕后,升至室温,搅拌2 h。
进行分相时,使用20 mL 20%碳酸钠溶液对有机相进行洗涤,然后合并水相。接着加入1 g活性炭进行脱色处理,持续0.5小时后进行抽滤,并用10 mL水进行洗涤。将滤液使用6 mol/L的盐酸调至pH=0.5~1,然后在0~5℃下搅拌析晶2小时。再次进行抽滤,进行少量水洗,最后在40℃下鼓风干燥,得到17.01 g浅黄色粗品,收率约为90.96%。
重结晶:将17 g 3-(氨甲酰甲基)-5-甲基己酸粗品加入到250 mL单口瓶中,加入85 mL乙酸乙酯和17 mL水,加热回流。之后,缓慢冷却至 0~5℃,析晶2 h,抽滤,用少量冷乙酸乙酯洗涤,20℃真空干燥,得到14.93 g类白色固体,m.p.为107~108℃,回收率约为87.82%。
(3)(±)-3-氨甲基-5-甲基己酸的合成
在250 mL反应瓶中,分别加入20 g氢氧化钠、90 mL水,配制成18%的氢氧化钠溶液。加入18.7 g 3-(氨甲酰甲基)-5-甲基己酸,呈浅黄色透明溶液。降温至-10℃以下,保持低温,缓慢滴加17.6 g溴素。
滴加完毕后,保持低温,搅拌反应1 h;升至 室温,搅拌反应1 h;在40℃温度下,搅拌反应1 h;在70℃温度下,搅拌反应1 h,停止反应,降至室温。
用盐酸调至p H=5.5~6.0,在温度为0~5℃下析晶2 h,抽滤,用少量冷水洗涤,40℃鼓风干燥,得到13.18 g白色固体,收率约为82.89%。
重结晶:将13.18 g 3-氨甲基-5-甲基己酸加入到250 mL单口瓶中,加入80 mL异丙醇和水(1∶1)的混合溶液,加热回流。之后,缓慢冷却至 0~5℃,析晶2 h,抽滤,用少量冷异丙醇水溶液洗涤,20℃真空干燥,得到12.7 g白色固体,m.p.为168~170℃,回收率约为96.36%。
参考文献:
[1]陈文华,祁秀秀,尤海烽. (±)-3-氨甲基-5-甲基己酸的合成 [J]. 中国医药工业杂志, 2019, 50 (07): 746-748. DOI:10.16522/j.cnki.cjph.2019.07.008
[2]张静. 普瑞巴林关键中间体3-氨甲基-5-甲基己酸的合成 [J]. 煤炭与化工, 2016, 39 (10): 73-75+148. DOI:10.19286/j.cnki.cci.2016.10.021
本文介绍了两种用于合成阿齐瑞格的方法,希望能为相关研究提供新的思路。
简介:RAGE 在多种疾病尤其是慢性病中起着重要作用,近年来RAGE抑制剂的研究比较热门,根据美国国家卫生研究所(National Institutes of Health)下属的药物临床试验网站(https://clinicaltrials.gov)检索的数据(截止至2020年7月),该靶点有57项研究处于临床研究阶段。
其中,有一个有机小分子化合物,称为阿齐瑞格(Azeliragon,曾用代号PF-04494700 或TTP488,CAS号:603148-36-3,化学名:[3-(4-{2-丁基-1-[4-(4-氯-苯氧基)-苯基]-1H- 咪唑-4-基-苯氧基)-丙基]-二乙基-胺),结构式见下图。2011年到了临床二期试验之后, 结构式才对外公开。已发表的文献不多,且集中在报道临床试验结果方面。
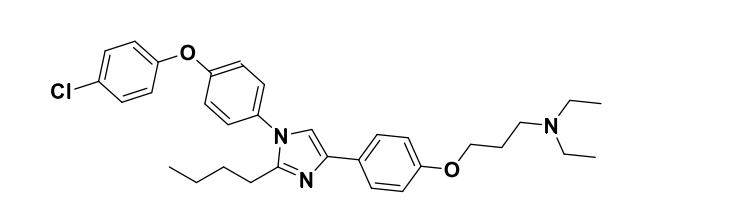
1. 研究概述:
作为针对AD的研究化合物,阿齐瑞格口服给药能够穿过血脑屏障,抑制RAGE与其配体Aβ1-42、S100、HMGB1及CML等的结合,同时也抑制sRAGE与Aβ1-42的结合。其作用机制涉及影响Aβ的积累、tau蛋白以及慢性炎症,从而减少神经元损伤。这有助于降低患者大脑认知损伤程度,延缓疾病的进展。
体外实验证实,阿齐瑞格能够有效抑制RAGE与多种配体(包括S100B、HMGB1和Aβ1-42)的结合。动物实验表明,小鼠在接受该药物后,其脾脏中Aβ1-42的聚集减少,IL-6和巨噬细胞集落因子的表达也有所降低。在APP过表达的转基因小鼠模型中,经过90天的阿齐瑞格治疗后,炎症因子(如TNF-α、TGF-β、IL-1)减少,中枢神经系统中Aβ的沉积减少,认知功能得到改善。
2. 合成
阿齐瑞格的结构较简单,分子中没有手性碳、磷、氮等不对称中心,不产生光学异构体。其合成方法报道很少,公开的文献主要为美国特兰斯泰克制药公司(Transtech Pharma, INC. USA)申请的2篇专利。
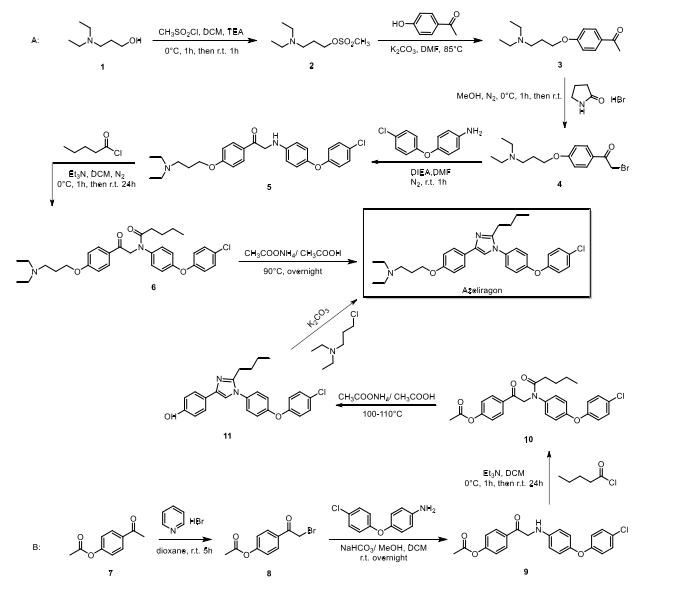
(1)2003年的专利WO2003075921合成方法见下图的路线A:以3-二乙基氨基丙醇1为原料,与甲磺酰氯通过醇解反应得到2,接着与4-羟基苯乙酮/碳酸钾发生取代反 应得3,与吡咯烷酮氢三溴化物在惰气保护下反应,酮基的α-H被溴取代生成4,再与 相等当量的4-氯苯氧基苯胺混合,在3当量的N, N-二异丙基乙基胺催化下生成5,接着 加入3当量的戊酰氯,以3当量的三乙胺为碱,反应得到6,最后在大过量的乙酸-乙酸 铵缓冲液中,90℃过夜反应,发生环合得到含咪唑环的目标产物。
(2)2011年的专利WO2011041198合成方法见下图的路线B:以4-乙酰基乙酸苯酯7为原料,加入三溴化吡啶鎓,酮基的α-H被溴取代生成8,接着与0.87当量的4-氯苯氧基苯胺混合,碳酸氢钠为碱,反应得到9,再加入1.5当量的戊酰氯,以2当量的 三乙胺为碱,反应得到10,在大过量的乙酸-乙酸铵缓冲液中,100-110℃过夜反应得到 11,最后再与3-二乙基氨基丙醇在碳酸钾作用下反应得到目标产物。
参考文献:
[1]谢集照.阿齐瑞格类似物合成和抗炎活性评价及青天葵多糖分离和抗炎活性评价[D].广西大学,2022.DOI:10.27034/d.cnki.ggxiu.2022.000010.
[2] Jones D, Gowda R B, Xie R. Substituted imidazole derivatives for treatment of Alzheimer's disease and synthesis and pharmacokinetics, WO2011041198A1 [P/OL]. 2011-.
[3] Mjalli A M M, Andrews R C, Gopalaswamy R, et al. Preparation of imidazole and benzimidazole derivatives that inhibit the interaction of ligands with RAGE, WO2003075921A2 [P/OL]. 2003-.
本文旨在探讨合成2-羟基-4-三氟甲基吡啶的方法。通过深入研究这一合成过程,有望为相关领域的发展提供新的见解和启发。
背景:啶磺草胺是美国陶氏益农公司开发的磺酰胺类内吸传导型、选择性冬小麦田苗后除草剂,具有杀草谱广、除草活性高、药效作用快等优点,广泛应用于世界各地。2-羟基-4-三氟甲基吡啶是合成啶磺草胺的关键中间体。
合成:
1. 方法一:
以5,5-二乙氧基-3-三氟甲基戊烯酸乙酯为原料,加入氨试剂一步环化实现了一锅法合成2-羟基-4-三氟甲基吡啶,该方法具有安全环保、反应条件温和、后处理方便、产品收率和纯度较高等优点,易于啶磺草胺的工业化生产,应用前景广阔。具体步骤如下:
在1L压力釜中加入5,5-二乙氧基-3-三氟甲基戊烯酸乙酯(142g,0.5mol),加入甲醇426g,降温至10℃,通入液氨(17g,1mol),然后关闭压力釜阀门,加热至100℃,保持压力0.1MPa,反应结束后,脱除90%的溶剂,加入142g水,过滤、烘干得73.8g 2-羟基-4-三氟甲基吡啶,收率90.6%,纯度99.2%。
2. 方法二:
S1、以乙烯基乙醚为起始原料,通过三氟乙酰化,得4-乙氧基-1,1,1-三氟-3-丁烯-2-酮;S2、将S1中得到的4-乙氧基-1,1,1-三氟-3-丁烯-2-酮与氯乙腈环合,得2-羟基-4-三氟甲基吡啶。
3. 方法三:
以乙烯基正丁醚为起始原料合成4-丁氧基-1,1,1-三氟-3-烯-2-酮,然后将该化合物与三甲基膦酰基乙酸酯反应得到中间体5-丁氧基-5-甲氧基-3-(三氟甲基)戊-2-烯酸甲酯及其异构体,中间体缩合成环得到化合物2-羟基-4-三氟甲基吡啶,具体步骤如下:
(1)4-丁氧基-1,1,1-三氟-3-烯-2-酮的合成
将70.1g乙烯基正丁醚(0.70mol)、55.40g吡啶(0.70mol)和100mL二氯甲烷加入到反应器中,搅拌降温到-10℃,然后开始滴加147.0g三氟乙酸酐(0.70mol),滴加过程中控制反应温度在-10℃~0℃,滴加结束后升温到25℃反应2小时。
反应结束后,将反应液冷却到-10℃,过滤反应产生的固体,少量二氯甲烷洗涤,滤液用200mL水洗涤、分液,少量二氯甲烷萃取,合并后用无水硫酸钠干燥,随后减压回收溶剂得到138.0g淡黄色液体,收率95.2%。
(2)5-丁氧基-5-甲氧基-3-(三氟甲基)戊-2-烯酸甲酯及其异构体的合成
在反应器中加入27.0g甲醇和23.7g三甲基膦酰基乙酸酯(0.13mol),搅拌降温到-10℃,氮气保护下滴加25.2g甲醇钠/甲醇溶液(30%,0.14mol),滴加过程中反应温度控制在-10℃~0℃,加完后搅拌5分钟,停止制冷,然后开始滴加反应(1)所得27.0g 4-丁氧基-1,1,1-三氟-3-烯-2-酮(0.13mol),加完后升温至25℃,反应4h。
反应结束后,减压回收溶剂甲醇,随后加入200mL水溶解,300mL石油醚萃取、分液,无水硫酸钠干燥后减压回收石油醚,得到34.8g红色液体产物,收率94.0%。
(3)2?羟基?4?三氟甲基吡啶的合成
将反应(2)所得20.00g 5-丁氧基-5-甲氧基-3-(三氟甲基)戊-2-烯酸甲酯及其异构体混合物(0.063mol)、22.00g醋酸铵(0.29mol)和20mL甲酰胺加入到反应器中,氮气保护下,升温到160℃反应8h。
反应结束后,降温到60℃,向反应体系加入20mL水和20mL饱和氯化钠溶液,搅拌一段时间后有固体析出,随后反应体系在-10℃冷却,过滤、真空干燥后得到8.45g淡黄色固体产物,收率74.0%。
[1]李立斌,李耀锋,张艳波,等. 2-羟基-3-氟-4-三氟甲基吡啶的合成工艺改进[J]. 山东化工,2022,51(5):11-13. DOI:10.3969/j.issn.1008-021X.2022.05.003.
[2] 江苏省农用激素工程技术研究中心有限公司,苏州大学. 一种2-氯-4-(三氟甲基)吡啶的合成方法:CN202310198178.0[P]. 2023-07-14.
[3] 湖南阿斯迪康药业有限公司. 一种2-氨基-4-三氟甲基吡啶的高效合成方法:CN202211077430.4[P]. 2022-11-25.
[4] 常州沃腾化工科技有限公司. 一种2-羟基-4-三氟甲基吡啶的制备方法:CN202211017938.5[P]. 2022-11-04.
本文旨在探讨利用5,6-二氢-3-(4-吗啉基)-1-(4-硝基苯基)-2(1H)-吡啶酮合成阿哌沙班的方法。通过深入研究这一合成过程,有望为相关领域的发展提供新的见解和启发。
背景:阿哌沙班是一种新型口服Xa因子抑制剂,是用于预防和治疗血栓的药品。5,6-二氢-3-(4-吗啉基)-1-(4-硝基苯基)-2(1H)-吡啶酮是合成阿哌沙班的重要中间体。
应用:合成阿哌沙班。
以5,6-二氢-3-(4-吗啉基)- 1-(4-硝基苯基)-2(1H)-吡啶酮和[(4-甲氧基苯基)肼基]氯乙酸乙酯为起始原料,依次经过[3+2]环合-消除、还原、酰胺化、环合、水解、与N,N'-羰基二咪唑(CDI)作用得活性酰胺中间体,再经氨水氨解得目标化合物阿哌沙班。具体步骤如下:
(1)1-(4-甲氧基苯基)-7-氧代-6-(4-硝基苯基)- 4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-羧酸乙酯(2)的制备
常温下(25℃),在三颈瓶中加入化合物1 (100 g,0.33 mol)、[(4-甲氧基苯基)肼基]氯乙酸乙酯(126.7 g,0.50 mol)、氯仿(1000 mL),搅拌,升温至61℃,缓慢滴加三乙胺(91.3 mL, 0.66 mol),61℃回流反应8 h,TLC监测反应结束。冰浴冷却至10℃,滴加3 mol·L-1 的稀盐酸溶液550 mL,反应2 h,TLC监测反应结束。反应液依次用水(100 mL×2)、饱和食盐水(100 mL×2)洗涤,无水硫酸钠干燥,过滤,蒸干,得到暗红色粘稠物。加入500 mL乙酸乙酯,搅拌30 min,析出大量黄色固体,过滤,依次用乙酸乙酯和水洗涤,得到黄色固体(2)108.2g,收率75.2%。
(2)1-(4-甲氧基苯基)-7-氧代-6-(4-氨基苯基)- 4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-羧酸乙酯(3)的制备
常温下(25℃),在三颈瓶中加入1000 mL 盐酸,搅拌下加入氯化亚锡(227.9 g,1 mol),温度稍下降后缓慢加入化合物2(108 g,0.25 mol),水浴下控温40~45℃,加料完毕,继续反应1 h。冷却降温至0℃,抽滤得白色固体。固体用热水 (100 mL)洗涤,抽滤,干燥,得到亮白色固体(3) 85.8 g,收率85.3%。
(3)1-(4-甲氧基苯基)-7-氧代-6-[4-(5-氯戊酰 胺基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡 啶-3-甲酸乙酯(4)的制备
常温下(25℃),在三颈瓶中加入化合物3 (85 g,0.21 mol)、三乙胺(58.3 mL,0.42mol)、 二氯甲烷(800 mL),搅拌30 min,使原料完全溶 解,冰浴降温至5℃,缓慢滴加5-氯戊酰氯 (32.1 mL,0.25 mol),控制反应温度在5~10℃,滴加完毕,继续搅拌30 min,TLC监测反应完全。 加入400 mL水,搅拌后静置分层,弃去水层,有 机层用水洗涤(100 mL×2),加入无水硫酸钠干 燥,抽滤,滤液减压浓缩,过滤,以正己烷洗涤滤 饼,得到黄色粉末(4)104.1g,收率94.5%。
(4)2.4 1-(4-甲氧基苯基)-7-氧代-6-[4-(2-氧代-1- 哌啶基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c] 吡啶-3-羧酸乙酯(5)的制备
常温下(25℃),在三颈瓶中加入化合物4 (104 g,0.20 mol)、氢氧化钠(24 g,0.6 mol)、甲丁基溴化铵(5 g)、二氯甲烷(1000 mL),搅拌30 min,升温至41℃,回流反应4 h,TLC监测反应 完成。加入300 mL水,静置分液,减压浓缩有机相,加入正己烷300 mL,冷却静置2 h。析出黄色粉末,过滤,滤饼用正己烷洗涤(100 mL×2),得到黄色粉末(5)87.8g,收率90.6%。
(5)1-(4-甲氧基苯基)-7-氧代-6-[4-(2-氧代哌 啶-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c] 吡啶-3-羧酸(6)的制备
常温下(25℃),在三颈瓶中加入化合物5 (87 g,0.18 mol)、水(400 mL)、四氢呋喃(400 mL),搅拌30 min,加入氢氧化钠溶液(21.4 g氢氧化钠溶于60 mL水),常温反应3 h(反应完毕时反应液为澄清状态)。加入8.7 g活性炭,40℃搅拌1 h,过滤,滤液冷却至10℃,滴加3 mol·L-1的盐酸溶液,调节p H值为2.0~3.0,搅拌1.5 h,析出大量固体。过滤,滤饼用水洗涤(100 mL× 2),得到黄色固体(6)70.8 g,收率86.3%。
(6)阿哌沙班(apixaban)的合成
在三颈瓶中加入化合物6(70 g,0.15 mol)和四氢呋喃(700 mL),分批加入N,N'-羰基二咪唑 (29.6 g,0.18 mol),搅拌30 min,升温至65℃反 应4 h(不再有气泡生成)。降温至10℃,加入 300 mL氨水,搅拌3 h,析出大量白色固体。过滤,依次用饱和碳酸钠溶液(50 mL×3)和乙醇 (50 mL×3)洗涤,得到白色固体59.6 g,收率 85.3%。
参考文献:
[1]张乾坤,沈思思,张峻铭等. 阿哌沙班的合成工艺研究 [J]. 中国药物化学杂志, 2022, 32 (03): 199-203. DOI:10.14142/j.cnki.cn21-1313/r.2022.03.005
[2]王鹏,林丽红. 阿哌沙班的合成工艺改进 [J]. 中国现代应用药学, 2019, 36 (14): 1783-1786. DOI:10.13748/j.cnki.issn1007-7693.2019.14.011
本研究旨在探讨合成与应用3-硝基-4-氯三氟甲苯的方法,希望通过这项研究为相关领域的合成化学和应用研究提供新的思路和实验支持。
背景:3-硝基-4-氯三氟甲苯(1)中的氯原子,由于受到硝基和三氟甲基这两个强吸电子基的影响,其活性较高,因此可以与多种亲核试剂发生反应。Richard在1971年报道了1与氢氧化钠在DMSO中生成2-硝基-4-三氟甲基苯酚的反应;陈德化等在1993年分别报道了1与乙二醇、3-硫杂-1,5-戊二醇、氨基乙醇进行的芳香亲核取代反应。此外,罗新湘等人在1998年和2000年相继报道了利用1合成出的几个含氟大杂环化合物,合成过程中经历了桥接、还原、环合等步骤。
1. 合成:
以对氯三氟甲苯和硝酸铵为原料,以离子液体为催化剂和溶剂,在常压下加热搅拌混合,完成对氯三氟甲苯的硝化反应;硝化反应完毕后,冷却静置,使反应体系分为液-液两相,上层为对氯三氟甲基苯的硝化产物4-氯-3-硝基三氟甲苯,下层为离子液体催化剂、铵盐、水的混合物;通过相分离移出上层产品,经洗涤、中和、真空除水后得到精制的4-氯-3-硝基三氟甲苯产品;下层含有水份的离子液体催化剂,经脱水、脱氨、干燥后,再次作为催化剂和溶剂,进行循环利用。
2. 应用:合成含氟化合物。
罗新湘等人以3-硝基-4-氯三氟甲苯(1)为起始原料,合成了新型含氟杂环化合物8和3个含氟中间体4,5,6。具体步骤如下:
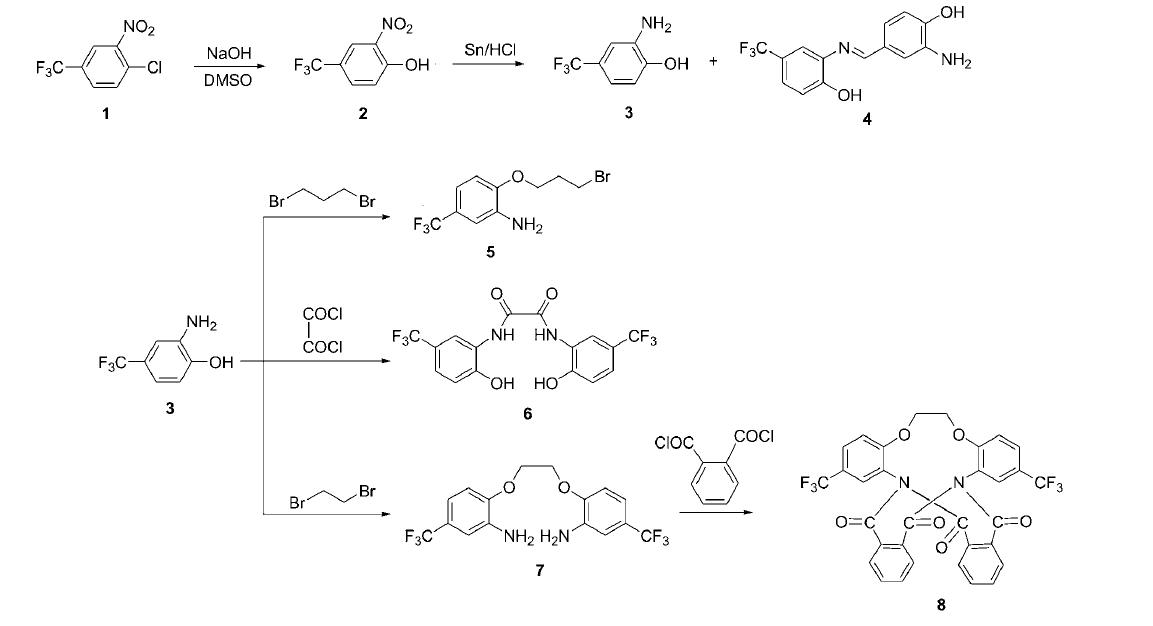
(1)化合物3,4的合成
在圆底烧瓶中加入2.60 g (0.126 mol)化合物2,30 g 锡粉和90 mL浓盐酸,加热使反应缓缓进行。升温至回流,待Sn粉反应完全,再回流1 h,冷却过夜,反应液中加入80 mL乙酸乙酯,用NaOH调节pH值至5~6,并不停搅拌,分出有机相,水层用乙酸乙酯萃取50 mL× 3,合并有机层,用无水Na2SO4 干燥,减压浓缩得固体,用乙酸乙酯/石油醚重结晶,得白色固体3 14.31 g。 母液浓缩,硅胶柱层析V(乙酸乙酯)/V(石油醚)=1/2,得灰色固体4 1.86 g。
(2)化合物5的合成
在圆底烧瓶中,加入1.77 g (0.01 mol)化合物3,100 mL丙酮,1.32 g (0.02 mol) KOH,60 ℃ 油浴,电磁搅拌下滴加4.40 g (0.02 mol) 1,3-二溴丙烷与20 mL丙酮的混合液,2 h滴完,滴完后继续反应3 h,冷却、过滤、浓缩滤液,硅胶柱层析[V(乙酸乙酯)/V(石油醚)=1/10]得固体5 0.93 g,产率31.3%. m.p. 34~35 ℃。
(3)化合物6的合成
在圆底烧瓶中加入100 mL无水苯,1.77 g (0.01 mol)化合物3,2.76 g (0.02 mol)无水碳酸钾,80 ℃油浴,电磁搅拌下滴加1.27 g (0.01 mol)乙二酰氯和10 mL无 水苯的混合液,0.5 h滴完,继续反应3 h,过滤,浓缩滤 液,得白色固体,用乙酸乙酯石油醚混合溶剂重结晶,得固体6 1.53 g,产率75%. m.p. 260~261 ℃。
(4)化合物8的合成
在圆底烧瓶中加入180 mL无水THF,加入少量钠片,回流3 h,加入0.2 g (0.53 mmol)化合物7,0.06 g (1.06 mmol) KOH,80 ℃油浴,电磁搅拌下滴加1.107 g (5.3 mmol)邻苯二甲酰氯和30 mL无水THF的混合液,3 h滴完,继续反应6 h,冷却、过滤、浓缩滤液,硅胶柱层析(氯仿)得固体8 0.23 g,产率70 %,m.p. 270~271 ℃。
参考文献:
[1] 江苏大华化学工业有限公司. 一种4-氯-3-硝基三氟甲苯的清洁制备方法. 2013-08-28.
[2] 罗新湘,黄筱玲,曲凡歧. 几个新型含氟化合物的合成[J]. 有机化学,2006,26(6):874-877. DOI:10.3321/j.issn:0253-2786.2006.06.022.
本文介绍了如何使用(E)-3-[3-(4-氟苯基)-1-异丙基吲哚-2-基]丙烯醛来合成氟伐他汀钠。
背景:(E)-3-[3-(4-氟苯基)-1-异丙基吲哚-2-基]丙烯醛是氟伐他汀钠的重要合成中间体。氟伐他汀钠是一种通过化学合成得到的降血脂新药,作为3-羟基-3-甲基戊二酰辅酶A(HMG-CoA)还原酶抑制剂,是继洛伐他汀、普伐他汀和辛伐他汀之后开发的第4个同类药物。氟伐他汀最早由瑞士Sandoz公司开发,在1993年投产并于1994年1月首次在英国上市,随后逐渐在美国、加拿大、新西兰等多个国家上市,并于1997年在我国注册。
氟伐他汀是一种强效降脂药物,无需代谢转化即具有药理活性。其分子中的氟苯吲哚部分模拟辅酶的结构,与HMG-CoA还原酶相互作用,竞争性地抑制该酶催化HMG-CoA转化为甲羟戊酸的关键步骤,从而减少细胞内胆固醇积累,增加低密度脂蛋白胆固醇(LDL-C)受体数量,恢复胆固醇代谢平衡,并促进LDL-C的清除。氟伐他汀为右旋和左旋两种消旋体组成,其中右旋活性强30倍。使用氟伐他汀能够降低血浆总胆固醇、甘油三酯和低密度脂蛋白,同时增加高密度脂蛋白水平,并抑制动脉血管粥样硬化的发展过程,通过抑制甲羟戊酸还原酶来抑制动脉肌细胞的增生和生长。
(E)-3-[3-(4-氟苯基)-1-异丙基吲哚-2-基]丙烯醛合成氟伐他汀钠的路线:
(1) 根据1988年美国专利4739073的公开内容,提供了一种合成(3R,5S)-氟伐他汀钠的方法。该方法以氟苯(4a)为起始原料,与氯乙酰氯反应形成4-氯乙酰氟苯(4b)。然后将化合物(4b)与氮异丙基苯胺(4c)进行缩合反应生成中间体(4d),再经过无水氯化锌催化下的环合反应形成吲哚环,得到化合物(4e)。接下来,将化合物(4e)与3-N-甲基-N-苯基胺基丙烯醛(4f)进行Vilsmeier-Haack反应,得到(E)-3-[3'-(4"-氟苯基)-1'-(1"-甲基乙基)吲哚-2'-基]-2-丙烯醛(4g)。然后,化合物(4g)与乙酰乙酸甲酯缩合生成β-羟基酮,随后使用三乙基硼烷和硼氢化钠作为还原剂将羰基还原为羟基,最后通过氢氧化钠的水解反应得到目标产物(I)(3R,5S)-氟伐他汀钠。
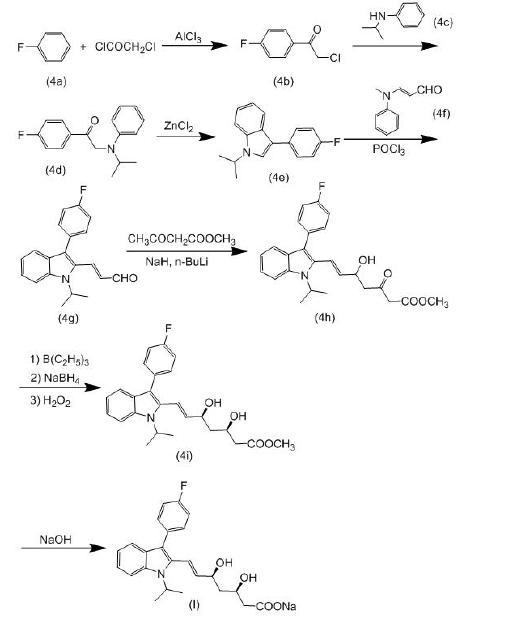
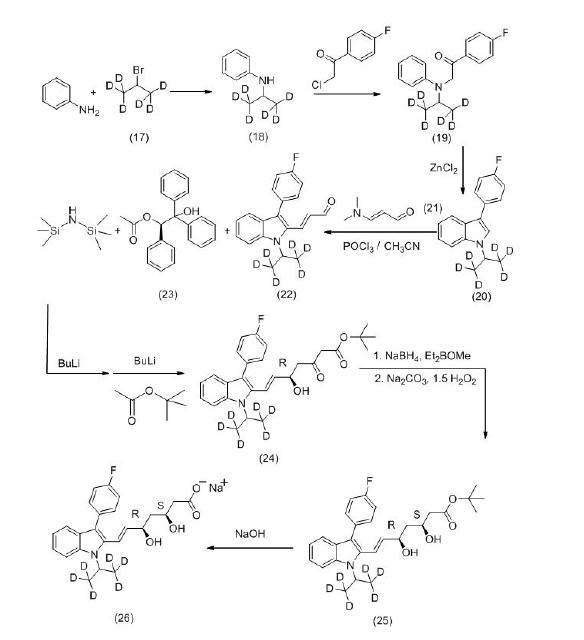
(2)以苯胺和含有6个氘原子的2-溴丙烷(17)为原料,生成N-异丙基苯胺-d6(18),再与4-氟-苯乙酰氯缩合生成化合物(19),经分子内环合生成化合物(20),再与N,N-二甲基 丙烯醛(21)发生Vilsmeier-Haack反应,生成( E ) -3-[ 3′-( 4″-氟苯基) -1′- (异丙基[d6])-吲哚-2′-基]-2-丙烯醛(22)。使用(R)-2-羟基-1,2,2-三苯基乙酸乙酯(23)作为手性催化剂,将中间体(22) 直接转化为5位具有所需要合成的R构型羟基的化合物(24)。使用syn-选择性还原剂 Et2BOMe/Na BH 4将化合物(24)中3位羰基选择性还原为羟基,直接得到所需要(3S,5R)构型的氟伐他汀叔丁酯-d6,经氢氧化钠水解得最终产物(3S,5R)-氟伐他汀钠-d 6,反应路线如图。
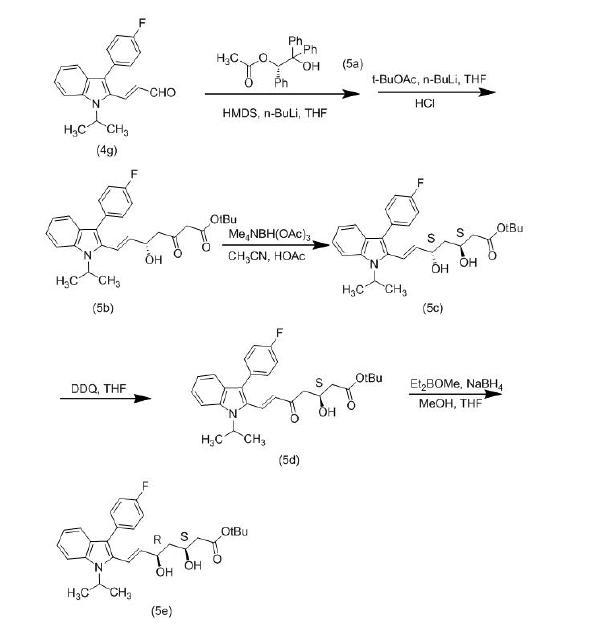
(3)根据Orin Tempkin等人于1997年在Tetrahedron发表的论文,研究人员采用了一种新的合成路线来制备(3S,5R)-氟伐他汀钠。首先,在合成中间体(4g)之后,使用手性催化剂(S)-2-羟基-1,2,2-三苯基乙酸乙酯(5a),将不饱和醛转化为具有相同构型的β-羟基酮酯,并与乙酸叔丁酯反应生成化合物(5b)。这个反应可以在同一个容器中进行,并具有高度的选择性。通过简单的重结晶处理,可获得约98%的对映体纯度。接下来,关键的中间体β-二酮可以经由以下步骤合成(3S,5R)-氟伐他汀钠。首先,研究人员使用抗选择性还原剂Me4NHB(OAc)3对化合物(5b)进行还原得到(3S,5S)-氟伐他汀叔丁酯(5c)。然后,他们使用DDQ将5位羟基氧化为酮(5d)。最后,通过使用syn-选择性还原剂Et2BOMe/NaBH4将5位还原为R构型的羟基,得到所需的(3S,5R)-氟伐他汀叔丁酯(5e)。通过简单的水解处理,即可得到目标化合物(3S,5R)-氟伐他汀钠。
参考文献:
[1]吴科颖. 氘标记药物标准品双氯芬酸、氟伐他汀钠的合成[D].南京航空航天大学,2010.
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手























