在医药化工中,S-三苯甲基-L-半胱氨酸是一种常见的中间体,用于多肽合成。为了保护巯基的高亲核性和易氧化性,常使用Fmoc作为保护基,得到Fmoc-S-三苯甲基-L-半胱氨酸。
Fmoc-S-三苯甲基-L-半胱氨酸通过氨基甲酸酯形成来保护胺基。它在酸性和氧化条件下相对稳定,但在温和的碱性条件下容易脱保护。它在肽固相合成方法中广泛应用,并可通过HPLC进行反应追踪。
将L-半胱氨酸盐酸盐和三苯基氯甲烷加入N,N-二甲基甲酰胺中,在适当条件下反应得到反应液。通过结晶和洗涤得到初品,再经过溶解、过滤和结晶得到S-三苯甲基-L-半胱氨酸。
将Fmoc-cl滴加到含有S-三苯甲基-L-半胱氨酸和碳酸氢钠的二氯甲烷溶液中,在适当条件下反应得到反应液。经过洗涤、干燥和旋干溶剂,最终得到Fmoc-S-三苯甲基-L-半胱氨酸。

图1 Fmoc-S-三苯甲基-L-半胱氨酸合成反应式
[1]CN 109134325 A
Fmoc-L-谷氨酰胺作为一种重要的氨基酸保护衍生物,在多肽合成领域发挥着举足轻重的作用。其独特的结构特征使其成为构建复杂肽链的理想起始物料,广泛应用于药物研发、生物材料制备等领域。
简介:
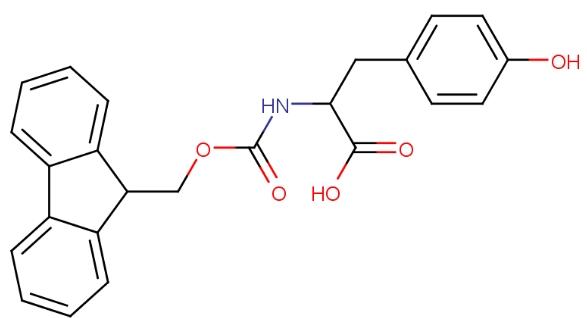
Fmoc-L-酪氨酸(Fmoc-Tyr-OH),分子式为C24H21NO5,具有一个手性中心,因此能够存在为两个对映异构体,并展现光学活性。作为酪氨酸的衍生物,Fmoc-Tyr-OH 在Fmoc固相肽合成中扮演了重要角色,是此类合成中常用的酪氨酸衍生物。这种化合物通常通过在水相中进行固相合成的方式制备。Fmoc-L-酪氨酸的结构如下图所示:
应用举例:
(1)制备胸腺五肽
王文琪等人报道了一种胸腺五肽新的制备方法,包括如下步骤:以羟甲基树脂为起始原料,以Fmoc保护的氨基酸为单体,第一个氨基酸采用Fmoc-Tyr-OH,以TBTU为缩合剂,逐个接上氨基酸,最后一个肽链采用Boc-Arg-OH;切肽,提纯,获得目标产物。该方法采用羟甲基树脂为固相载体,以Fmoc保护的氨基酸为单体,第一个氨基酸采用Fmoc-Tyr-OH,不需要侧链保护,以TBTU为缩合剂,逐个接上氨基酸,每步接肽收率≥98%,最后一个肽链采用Boc-Arg-OH,降低了生产成本,简化工艺,用2NHCl/HAc切肽,加乙酸乙酯沉淀粗品,条件温和,三废污染少。
(2)合成含 Tyr(P) 肽的新方法
JohnW.Perich等人描述了一种高效合成含 Tyr(P) 肽的新方法,即在酪氨酸残基被掺入肽链后对其进行“亚磷酸三酯”磷酸化。在这种方法中,肽树脂通过 Fmoc 固相肽合成 (PyBOP® 偶联) 组装,其中酪氨酸残基被掺入为 Fmoc-Tyr-OH。然后用二叔丁基 N,N-二乙基亚磷酰胺/1H-四唑对 N 端酪氨酸肽树脂进行磷酸化,中间体二叔丁基亚磷酸三酯通过间氯过氧苯甲酸处理而氧化。然后在通常条件下继续肽合成,并通过酸解处理对肽树脂进行脱保护。通过 IVPNY(P)VEE 和 Fcγ 受体肽 EAENTITY(P)SLLKHPEAL 的合成证明了“在线”磷酸化方法的效率提高,产量都很高。
(3)制备基于碳纳米材料的氨基酸保护基水凝胶
由低分子量胶凝剂自组装形成的分子凝胶因其在药物输送中的潜在应用而受到越来越多的关注。特别是,肽和氨基酸自发自组装成三维纤维网络的能力已被用于水凝胶的开发。在此背景下,Guilbaud-Chereau等人研究了芳香族氨基酸衍生物二元混合物形成水凝胶的能力。碳纳米材料,即氧化碳纳米管或氧化石墨烯,被掺入两种最稳定的水凝胶中,分别由 Fmoc-Tyr-OH/Fmoc-Tyr(Bzl)-OH 和 Fmoc-Phe-OH/Fmoc-Tyr(Bzl)-OH 形成。水凝胶的形成主要由芳香相互作用驱动。最后,将亲水性模型药物(L-抗坏血酸)以高浓度负载于混合水凝胶中,在近红外光照射下,碳纳米材料产生的热量引发大量药物释放,为药物控制输送提供了有趣的前景。
(4)植物硫肽素活性中心液相合成
作为一种植物多肽激素,PSK对植物的生长增殖起重要的调节作用。其核心结构[H-Tyr(SO3H)-Ile-Tyr(SO3H)-OH]具有20%的PSK生物活性。由于PSK在植物体内含量低,提取纯化困难,固相合成产物的产率低,李琳等人采用经典的液相方法合成其核心结构。Tyr和Ile的氨基Fmoc保护在冰浴反应后,室温分别反应5h和4h达到最高产率,液相色谱检测纯度分别为90.5%和97.6%。酪氨酸的羧基叔丁基保护反应通过实验室自制异丁烯气体进行保护,按照最优反应原料比例室温反应4d产率达到65.7%,液相色谱分析纯度达到92.1%。
参考:
[1] YI YANG, LENA HANSEN, PER RYBERG. Side-Chain Unprotected Fmoc-Arg/His/Tyr-OH Couplings and Their Application in Solid-Phase Peptide Synthesis through a Minimal-Protection/Green Chemistry Strategy[J]. Organic process research & development,2022,26(5):1520-1530. DOI:https://doi.org/10.1021/acs.oprd.2c00083.
[2] 李琳. 植物硫肽素活性中心液相合成及抑制生菜衰老活性研究[D]. 湖北:华中农业大学,2011. DOI:10.7666/d.Y2003836.
[3] JOHNW.PERICH. Synthesis of Tyr(P)-containing peptides via ‘on-line’ phosphorylation of the tyrosine residue on the solid phase[J]. Letters in Peptide Science,1996,3(3):127-132. DOI:10.1007/BF00132975.
[4] GUILBAUD-CHEREAU, CHLOE, DINESH, BHIMAREDDY, SCHURHAMMER, RACHEL, et al. Protected Amino Acid-Based Hydrogels Incorporating Carbon Nanomaterials for Near-Infrared Irradiation-Triggered Drug Release[J]. ACS applied materials & interfaces,2019,11(14):13147-13157. DOI:10.1021/acsami.9b02482.
[5] 上海苏豪逸明制药有限公司,周逸明,崔颀. 一种胸腺五肽的制备方法:CN201310335626.3[P]. 2013-10-16.
辛卡利特是一种重要的胆囊素八肽,其制备方法备受研究人员的关注。
简介:辛卡利特是一种人工合成的胆囊素八肽,主要用于促胆囊收缩,胆囊诊断试剂,具有胆囊收缩素的全部生物活性。研究表明辛卡利特具有减轻肺脏间质水肿及白细胞浸润,抑制促炎细胞因子生成,改善肺组织结构损伤,改善血流动力学障碍,延缓平均动脉压下降和肺动脉压升高,改善肝肾微循环血流,回复心率,降低肺动脉高压,改善潮气量等作用。临床上可以用来治疗慢性胰腺炎,内毒素血症,内毒素休克,脓毒血症,多器官功能障碍综合症,全身性炎症反应综合征等。
合成:辛卡利特的制备方法主要有酶法和化学法两种。其中,酶法合成由于涉及酶制剂种类比较多、酶制剂价格昂贵,且中间产物分离困难、制备周期长,目前已不再使用酶法制备辛卡利特。化学合成是目前常用的辛卡利特制备方法。CN201410105423.X公开了一种制备辛卡利特的方法,该方法中选择了氨基树脂,在低温条件下用三氟乙酸裂解多肽树脂,虽然能减少磺酰基脱落,但是不能完全避免,而且会导致多肽裂解不完全,降低粗品收率;CN200810043920.6公开了一种制备辛卡利特的方法,该方法选择用氨水裂解多肽树脂,其中氨水有毒,对眼、鼻、皮肤有刺激性和腐蚀性,不利于后期产业化,而且选用的Fmoc-Asp
(odmab)-OH这一原料价格昂贵,增加了生产成本,没有显著的经济效益。CN201810365226.X公开了一种制备辛卡利特的方法,该方法也是选取了两肽片段树脂作为起始树脂,但是Asp1选择了Fmoc-Asp (Ome)-OH保护策略,在裂解后还需要水解去除Ome。
1. 专利CN 110317257 A发明了涉及一种辛卡利特固液相结合的合成方法,主要解决现有技术存在的原料昂贵以及后处理繁琐的技术问题。具体步骤为:
(1)Fmoc-Asp(Otbu)-Osu,NH 2 -Phe-CONH 2 溶于四氢呋喃中,加入DIEA,搅拌反应2小时,旋蒸除去四氢呋喃,加入体积比95%TFA溶液搅拌反应1-2小时,去除叔丁基,再加乙醚结晶得到Fmoc-Asp- Phe-CONH 2 二肽片段;
(2)将二氯三苯基氯树脂置于圆底烧瓶中,加入Fmoc-Asp-Phe-CONH 2 二肽片段,加入二氯甲烷,DIEA,搅拌反应2小时,再加入甲醇震荡反应30分钟,过滤除去滤液;用二氯甲烷洗涤树脂三次,干燥,得到Fmoc-Asp(β-2Cl-Trt Cl 树脂)-Phe-CONH 2 ;
(3)将Fmoc-Asp(β-2Cl-Trt Cl 树脂)-Phe-CONH 2 置于多肽反应器中,加入脱保护试剂,氮气搅拌反应30分钟,抽干,再用DMF洗涤5次,加入相应的保护氨基酸,HBTU/NMM为缩合剂,DMF做溶剂,反应30-60分钟,茚三酮检测反应终点,反应结束后抽真空排干反应液,用DMF洗涤3次,再加入脱保护试剂,依次循环,直至连完最后一个天冬氨酸,脱保护,得到辛卡利特树脂肽;
脱保护试剂为体积比20%哌啶/DMF溶液,反应中所提到保护氨基酸依次是Fmoc-Met-OH,Fmoc-Trp-OH,Fmoc-Gly-OH,Fmoc-Met-OH,Fmoc-Tyr(SO 3 h)-OH,Fmoc-Asp(Ofm)-OH;
(4)将辛卡利特树脂肽置于圆底烧瓶中,加入裂解试剂,常温条件下反应1小时,抽滤滤去树脂颗粒,收集滤液,减压浓缩旋去溶剂,得到辛卡利特中间体;再经高液相色谱分离纯化、冻干后得到辛卡利特精品。
2. CN 108059665 B发明了一种辛卡利特的制备方法,主要解决现有酶法制备存在的试剂难以去除以及化学法存在的产品性能不稳定、收率不理想的技术问题。该方法包括如下步骤:(1)将固相合成所得的粗肽用氨水充分溶解,过超滤柱;(2)连接超滤柱与高压单泵,选用醋酸水作为淋洗剂,得到去TFA盐的辛卡利特粗品中间液体;(3)对去TFA盐的辛卡利特粗品中间液体用NH 4 HCO 3 溶液调节pH值为7.25;(4)将辛卡利特粗品中间液体,用固定相为十八烷基硅烷键合硅胶的反相硅胶柱,流动相为A相0.2%醋酸水溶液与B相乙腈溶液,纯化得到高纯的醋酸盐形式的辛卡利特液体;(5)将得到的醋酸盐形式的辛卡利特液体浓缩冻干,最终得到含量超过98.5%的固体辛卡利特。
参考文献:
[1] 吉尔生化(上海)有限公司. 一种辛卡利特的制备方法:CN201711282755.5[P]. 2021-05-18.
[2] 吉尔生化(上海)有限公司. 一种辛卡利特的固液相合成法:CN201910474762.8[P]. 2019-10-11.
布舍瑞林是一种具有广泛药理活性的重要化合物,其多种合成方法的研究对于推动该药物的生产和应用具有重要意义。
简介:布舍瑞林由西德赫斯特公司(Hoechst AG,现已并入赛诺菲)开发,1984年在西德首次上市。布舍瑞林是根据促性腺激素释放激素(GnRH)改造的多肽类药物。布舍瑞林可促使黄体生成素(Luteinizing Hormone,LH)、促卵泡激素(Follicle Stimulating Hormone,FSH)和性激素的分泌增加,其促进LH和FSH释放的作用效力分别为天然促性腺激素释放激素的19倍和26倍,长期使用引起垂体机能下调,导致LH和FSH的分泌减少。
合成:
1. 目前国内报道布舍瑞林的制备方法主要有:(1)片段缩合法:在液相采用碳二亚胺法将氨基酸偶联,采用固相合成Pyr-His-Trp-Ser-Tyr-OH,与D-Ser(tBu) -Leu-Arg-Pro-固相支撑物缩合得到布舍瑞林;固相合成片段Pyr-His(Trt)-Trp-Ser(Trt)-Tyr(Bzl)-D-Ser(tBu)-Leu-OH,液相合成H-Arg-Pro-NHEt 两片段在液相体系中缩合得到布舍瑞林。存在步骤繁琐、合成周期长,在片段缩合时位阻大、效率不高、且侧链基团裸露、引发副反应,造成粗肽纯度和收率较低的问题。(2)逐一合成法:在液相体系中,以H-Pro NH-C2H5为起点,加入氨基酸衍生物Z-Arg(Z)-OH、以DLL和HOBT为催化剂反应,然后按照顺序分别偶联剩余的氨基酸;采用HMPB-MBHA、2-CTC 树酯和不同氨基酸原料固相合成全保护肽,经乙胺化、脱保护得到布舍瑞林。使用的Fmoc-Tyr-OH侧链羟基不带保护,在缩合时很容易发生酯化副反应; Fmoc-His-OH不带保护基,咪唑基易发生N-酞胺化,且产物易产生消旋,降低了反应收率。
2. 日本伊藤株式会社公布的专利JP2006169165中提供了一种微通道反应器系统合成多肽的方法,其中提到利用该装置进行布舍瑞林片段缩合的操作方法。该方法中将三肽片段pGlu-His-Trp-OH和六肽片段H-Ser-Tyr-D-Ser(tBu)-Leu-Arg-Pro-NHEt以 HBTU/DIPEA作缩合剂,33%NMP/DMF作溶剂条件下缩合得到布舍瑞林粗肽,但未提及这两个片段具体的合成方法及收率等相关数据。就片段缩合工艺而言,由于两个片段中多个氨基酸侧链未保护,在片段缩合时必然会产生很多杂质,使得粗品纯度和收率低。
3. 以色列TEVA制药公司公布的专利WO2006/119388 A2 中布舍瑞林的制备工 艺如图,具体包括以下步骤:1,使用2-CTC树脂与Fmoc-Pro-OH制备得到 Fmoc-Pro-CTC 树脂,用固相法合成Pyr-His-Trp-Ser-Tyr(Bzl)-D-Ser(tBu)-Leu-Arg( NO2)-Pro-CTC树脂;2,使用1%TFA/DCM溶液裂解全保护肽得到中间体 Pyr-His-Trp-Ser-Tyr(Bzl)-D-Ser(tBu)-Leu-Arg(NO2)-Pro-OH;3,将中间体在乙胺/DMF 溶液中通过缩合剂缩合得到Pyr-His-Trp-Ser-Tyr(Bzl)-D-Ser(tBu)-Leu-Arg(NO2)-Pro-NHEt;4,将步骤3所得布舍瑞林全保护肽进行氢化脱除Bzl及NO2保护基,并纯化得到布舍瑞林。该工艺的优点是步骤简单,合成效率高。缺点是在用固相法合成中间体片段时,使用了单保护的Fmoc-Ser-OH,在进行缩合反应时,其裸露的侧链羟基会引起副反应;另外还使用了Fmoc-His(Fmoc)-OH,该保护氨基酸不稳定,市场上难以获得高纯度的该氨基酸原料,不利于规模化生产,且当Fmoc保护基脱除后咪唑基有反应活性,在缩合下一位氨基酸时会带来副反应,造成粗肽纯度低,影响合成收率。

4. 西班牙Lipotec公司发布的专利EP1179537A1中布舍瑞林的制备工艺如图,具体包括如下步骤:1,将Fmoc-Pro-OH通过DIPEA连接到2-CTC树脂上,用固相法合成Pyr-His(Mmt)-Trp-Ser(Trt)-Tyr(2-ClTrt)-D-Ser(tBu)-Leu-Arg-Pro-2-CTC 树脂;2,使用AcOH/TFE/DCM=1/2/7(V/V/V)或含1~30%醋酸的有机溶剂将中间体肽链从树脂上裂解下来,得到Pyr-His(Mmt)-Trp-Ser(Trt)-Tyr(2-ClTrt)-D-Ser(tBu)-Leu-Arg-Pro-OH;3,再用<5%TFA溶液脱除Mmt、Trt及2-ClTrt侧链保护基得到 Pyr-His-Trp-Ser-Tyr-D-Ser(tBu)-Leu-Arg-Pro-OH;4,将步骤三所得中间体与NH2Et 在缩合剂条件下缩合得到布舍瑞林粗肽,再经过反相HPLC纯化得到布舍瑞林纯品, 总收率为38~45%。该工艺与TEVA公司专利WO2006/119388A2有类似之处,都是以Fmoc-Pro-CTC树脂为起始合成中间体片段,再在液相中进行C末端乙胺化修饰。不同的是Lipotec公司的专利中使用了不同的保护氨基酸如Fmoc-His(Mmt)-OH 及Fmoc-Tyr(2-ClTrt)-OH,为合成布舍瑞林提供了更多的路线选择。但与此同时,由于这两个保护氨基酸不常用,市场价格昂贵,在规模化生产时不利于降低成本。另外,由于中间体肽链中His、Tyr及Ser侧链基团裸露,在缩合乙胺时会产生相应杂质,更好的方法应该是缩合乙胺后再脱除侧链保护基。

参考文献:
[1]赵红玲,高杨,代涛等.布舍瑞林合成工艺的优化[J].中国抗生素杂志,2016,41(04):285-288.DOI:10.13461/j.cnki.cja.005709.
[2]陆冬冬. 抗肿瘤多肽药布舍瑞林的合成研究[D].苏州大学,2014.
[3]琚滢蓥,徐宏伟,叶小惠等.布舍瑞林的研究概述[J].海峡药学,2014,26(04):12-14.
组氨瑞林是一种常用的药物,具有抗过敏和抗组胺作用,并被广泛应用于治疗过敏性疾病。本文将介绍如何通过合成方法来制备组氨瑞林。
简介:组氨瑞林是一种促黄体激素释放激素(LHRH)激动剂,美国学者萧尔(Shore)等报告,对于晚期前列腺癌患者,每年一次给予组氨瑞林长效埋植剂皮下埋植,能长期稳定地抑制睾酮,使其保持去势水平。
现有的合成方法均存在着一定得缺点,每一次脱保护均需用三氟乙酸,故三废污染多,接脑收率低,生产成本高 ,限制了组氨瑞林的大规模生产和应用。
合成:
1. 专利CN 113603751 A 提供一种全液相合成组氨瑞林的方法,涉及医药技术领域。包括如下步骤:采用液相法合成化合物1Fmoc-Trp(Boc)-Ser(tBu)-Tyr(tBu)-OH和化合物2R1-D-His(Bzl)-Leu-OR2 ,化合物2经脱保护后与化合物1缩合得到化合物4,化合物4经脱保护再分别接入-His(R 3 )-、R4 -Pyr-片段得到化合物7,化合物7经皂化后接入-Arg(pbf)-Pro-NHEt片段得到化合物10,化合物10经裂解后得到组氨瑞林粗品。该发明提供方法制得的组氨瑞林纯度可达80%以上。具体如下:
S1、化合物1:Fmoc-Trp(Boc)-Ser(tBu)-Tyr(tBu)-OH;
S2、化合物2:R1-D-His(Bzl)-Leu-OR2 ;
S3、化合物3:H-D-His(Bzl)-Leu-OR2 ;
S4、化合物4:Fmoc-Trp(Boc)-Ser(tBu)-Tyr(tBu)-D-His(Bzl)-Leu-OR2;
S5、化合物5:H-Trp(Boc)?Ser(tBu)-Tyr(tBu)-D-His(Bzl)-Leu-OR2 ;
S6、化合物6:H-His(R3 )-Trp(Boc)-Ser(tBu)-Tyr(tBu)-D-His(Bzl)-Leu-OR2 ;
S7、化合物7:R4 -Pyr-His(R)-Trp(Boc)-Ser(tBu)-Tyr(tBu)-D-His(Bzl)-Leu-OR2 ;
S8化合物8:R4-Pyr-His(R3)-Trp(Boc)-Ser(tBu)-Tyr(tBu)-D-His(Bzl)-Leu-OH;
S9、化合物9:H-Arg(pbf)-Pro-NHEt;
S10、化合物10:R4 -Pyr-His(R3)-Trp(Boc)-Ser(tBu)-Tyr(tBu)-D-His(Bzl)-Leu-Arg(pbf)-Pro-NHEt;
S11、组氨瑞林粗品的制备;
其中,R1为氨基保护基团,包括Fmoc、Z、Boc中的任意一种;R2为羧基保护基团,包括甲Me,乙酯Et,苄酯Bzl,三苯甲酯Tr中的任意一种;R3包括Boc或Trt中的任意一种;R4为氨基保护基团,包括Fmoc、Z、Boc中的任意一种。
2.专利CN 114315978 A提供一种固液合成组氨瑞林的方法属于医药合成技术领域。该方法包括以下步骤:S1、用固相法合成化合物1:Boc-Pyr-His(Boc)-Trp(Boc)-Ser(tBu)-Tyr(tBu)-OH;S2、用液相法合成化合物2:D-His(Bzl)-Leu-OMe;S3、用液相法合成化合物3:H-Arg(pbf)-Pro-NHEt;S4、在液相中以化合物1和化合物2为反应单元合成化合物4:S5、在液相化合物4皂化合成化合物5;S6、液相法以化合物3和化合物5为反应单元合成化合物6;S7、化合物6去侧链保护基团得组氨瑞林粗品。该发明提供的方法合成的组氨瑞林粗品纯度可达85%以上。具体为:
S1、用固相法合成化合物1:Boc-Pyr-His(Boc)-Trp(Boc)-Ser(tBu)-Tyr(tBu)-OH;
S2、用液相法合成化合物2:D-His(Bzl)-Leu-OMe;
S3、用液相法合成化合物3:H-Arg(pbf)-Pro-NHEt;
S4、在液相中合成化合物4:Boc-Pyr-His(Boc)-Trp(Boc)-Ser(tBu)-Tyr(tBu)-D-His(Bzl)-Leu-OMe;
S5、在液相中合成化合物5:Boc-Pyr-His(Boc)-Trp(Boc)-Ser(tBu)-Tyr(tBu)-D-His(Bzl)-Leu-OH;
S6、液相法合成化合物6:Boc-Pyr-His(Boc)-Trp(Boc)-Ser(tBu)-Tyr(tBu)-D-His(Bzl)-Leu-Arg(pbf)-Pro-NHEt;
S7、合成组氨瑞林粗品:Pyr-His-Trp-Ser-Tyr-D-His-Leu-Arg-Pro-NHEt。
3. 专利CN 102850437A发明了涉及组氨瑞林的一种合成方法,主要解决现有的组胺瑞林合成方法接肽收率低,三废污染多的技术问题。该发明的技术方案 :一种组氨瑞林的合成方法,包括如下步骤 :a.以CTC 树脂为起始原料,在碱性条件下与 Fmoc-Pro-OH的羧基相连,得到 Fmoc-Pro-CTC 树脂;b.其余8个氨基酸按照组氨瑞林依次合成 ;c.全保护切肽 :用全保护切割试剂裂解得 :yr-His(Trt)-Trp(Boc)-Ser(tbu)-Tyr(tbu)-D-His(Bzl)-Leu-Arg(pbf)-Pro9肽片段 ;d.用乙胺与全保护 9 肽片段在HOBT的作用下获得全保护组氨瑞林片段;e.裂解全保护组氨瑞林片段获得组氨瑞林粗品。
参考文献:
[1] 湖南三太药业有限公司. 一种固液合成组氨瑞林的方法:CN202111662600.0[P]. 2022-04-12.
[2] 湖南三太药业有限公司. 一种全液相合成组氨瑞林的方法:CN202111006173.0[P]. 2021-11-05.
[3] 上海吉尔多肽有限公司,吉尔生化(上海)有限公司,滨海吉尔多肽有限公司. 一种组氨瑞林的合成方法:CN201210334868.6[P]. 2013-01-02.
地洛瑞林是一种重要的化合物,其制备方法备受研究人员关注。本文将介绍两种地洛瑞林的制备方法,为研究人员提供参考。
简介:地洛瑞林又称 LHRH-Hydrogel implant,RL0903。地洛瑞林是一种人工合成的九肽,用于治疗中枢性性早熟症。地洛瑞林是一种促黄体激素释放激素(LHRH) 激动剂,美国学者 Shore 等报告,对于晚期前列腺癌患者,每年一次给予地洛瑞林长效埋植剂皮下埋植,能长期稳定地抑制睾酮,使其保持去势水平。
现有的合成方法均存在着一定的缺点,每一次脱保护均需用三氟乙酸,故三废污染多,接肤收率低,生产成本高,限制了地洛瑞林的大规模生产和应用。
合成:
1. 专利CN 104004054 A发明了一种地洛瑞林的合成方法,即以2-氯三苯甲基氯树脂为原料,在碱性条件下与芴甲氧羰基保护脯氨酸的羧基相连,得到芴甲氧羰基保护脯氨酸-2氯三苯甲基氯树脂,再通过接肽反应依次接入地洛瑞林剩余氨基酸,用全保护切割试剂裂解获得全保护9肽片段,再通过乙胺化获得全保护地洛瑞林片段,最后裂解全保护地洛瑞林片段获得地洛瑞林粗品。
步骤如下 :
(1)制备芴甲氧羰基保护脯氨酸-2氯三苯甲基氯树脂;
(2)在步骤(1)所得的芴甲氧羰基保护脯氨酸-2氯三苯甲基氯树脂上依次接入地洛瑞林的剩余氨基酸,获得 9肽 树脂;
(3) 用全保护切割试剂裂解步骤(2) 所得的9 肽树脂,获得全保护 9肽片段;
(4)乙胺化步骤(3) 所得全保护 9 肽片段,获得全保护地洛瑞林片段;
(5)裂解步骤(4) 所得全保护地洛瑞林片段,获得地洛瑞林粗品。
该发明通过固液结合合成地洛瑞林收率可达 84.1%。同时,避免了纯固相合成从树脂上切割不安全,不方便,缩短了合成时间,简化了纯液相片段合成的繁琐,使生产成本大大降低适于产业化推广。
2. 地洛瑞林的合成方法主要是固相合成。固相合成中需要使用大量的昂贵的多肽树脂,这给企业的大规模生产带来了成本的压力,不仅如此,在最后切肽的工序中,叔丁基很容易在酸性条件下被脱除,将生成其他杂质产物。
从现有的各种资料研究状况来看,液相合成地洛瑞林是现阶段各大公司和学校科研机构的主要方法。例如:国内专利 CN201410184663.3 采用了固液相结合的方法合成地洛瑞林,但是相对时间较长、固相合成杂质较多、纯化难度较高、受到固相设备的限制,产业化放大也存在问题。
专利CN 105254701A发明了一种地洛瑞林的合成方法,所述的地洛瑞林Pyr-His-Trp-Ser-Tyr-D-Trp-Leu-Arg-Pro-NHEt 是由五片段Pyr-His-Trp-Ser-Tyr-OH与四肤片段D-Trp-Leu-Arg-Pro-NHEt 在缩合剂的存在下缩合而成。本发明采用5+4的片段合成法,可直接将五脑片段 Pyr-His-Trp-Ser-Tyr-OH 和四脑片段D-Trp-Leu-Arg-Pro-NHEt 在缩合剂的存在下缩合成地洛瑞林。本方法不但缩短了合成周期、避免了传统方法中苛刻的反应条件,而且收率高、产品纯度好、成本低、反应条件温和、适合于工业化生产。
所述的地洛瑞林 Pyr-His-Trp-Ser-Tyr-DTrp-Leu-Arg-Pro-NHEt 是由五肤片段 R1-Pyr-His(R2)-Trp(R3)-Ser(R4)-Tyr(R5)-0H与四肤片段 R6-D-Trp(R3)-Leu-Arg(R7)-ProNHEt 在缩合剂的存在下缩合、脱保护而制成;
其中:
R1 选自 BOC、Z、Fmoc或H;
R2 选自BOC、Z、Bzl、Trt、Tos、Bom、Dnp 或H;
R3 选自 BOC、For、H或Z;
R4 选自 Bzl、H、Tbu、Z、Boc 或 Tos ;
R5 选自 Bzl、H、Tbu、2,6-di-C1-Bzl、Me、乙或 2-Cl-Z;
R6 选自 BOC、Z或Fmoc ;
R7 选自 BOC、(BOC)2、PBF、TOS、NO2、H、PMC、HCL、ADOC 或Mts。
参考文献:
[1] 江苏诺泰生物制药股份有限公司. 一种德舍瑞林的合成方法:CN201510735214.8[P]. 2016-01-20.
[2] 安徽瀚海博兴生物技术有限公司. 一种徳舍瑞林的合成方法:CN201410184663.3[P]. 2014-08-27.
背景及概述[1]
Fmoc-L-缬氨酸是一种氨基酸衍生物,可以通过L-缬氨酸与氯甲酸-9-芴基甲酯反应制备得到。有研究表明,Fmoc-L-缬氨酸可用于合成伐昔洛韦。


取500mL的反应瓶,分别向反应瓶中加入手性氨基酸S-3a(4.5g,38.8mmol)、二氧六环(40mL)和10%碳酸钠(100mL),将反应瓶置于冰浴中,机械搅拌,向滴液漏斗中加入氯甲酸-9-芴基甲酯(10.0g,38.8mmol)和二氧六环(100mL),缓慢滴入反应瓶,逐渐恢复至室温并搅拌过夜。反应完成后加水100mL,用50mL乙醚萃取三次,取水相放入冰浴中冷却,加1M稀HCl至PH为1。水溶液用50mL乙酸乙酯萃取三次。油相合并后用硫酸镁干燥,过滤旋干后得到中间体S-4a,即Fmoc-L-缬氨酸(12.6g,96%)。白色固体.1H NMR(400MHz,CDCl3)δ7.77(d,J=7.2Hz,2H),7.60(d,J=6.0Hz,2H),7.36(dt,J=34.8,7.2Hz,4H),5.28(d,J=8.8Hz,1H),4.42(d,J=6.8Hz,2H),4.36-4.33(m,1H),4.24(t,J=6.8Hz,1H),2.27-2.21(m,1H),1.01(d,J=6.4Hz,2H),0.95(d,J=6.8Hz,2H).
将L-缬氨酸为0.94克(0.008摩尔)固体溶于10%碳酸钠溶液中,搅拌使L-缬氨酸固体充分溶解,在20~30℃,滴加溶于甲苯(2~205毫升)地 9-芴甲氧羰基氯(2.10克,0.008摩尔)溶液,滴加30~60分钟,滴加 结束,在20~30℃搅拌1~8小时,加30~200毫升水稀释,用乙酸正丁 酯(80毫升)萃取除去过量的9-芴甲氧羰基氯,所得水相用浓盐酸酸 化至PH=0.5~3.5,然后用乙酸正丁酯(80毫升)萃取,所得油相用水 洗涤除去盐酸,油相浓缩除去乙酸正丁酯溶剂,白色结晶析出,过滤,干 燥得产物Fmoc-L-缬氨酸2.36克,得率为86.9%,熔点:143~144℃。
CN200710092592.4报道了被保护的伐昔洛韦,即N-(9-芴甲氧基羰基)-L-缬氨酸2-[(2-氨基-1,6-二氢-6-氧代-9H-嘌呤-9-基)甲氧基]乙酯及其制备方法。本发明还涉及一种制备伐昔洛韦的方法,所述方法包括以下步骤:用偶联剂使FMOC-L-缬氨酸与阿昔洛韦发生偶联,形成被保护的伐昔洛韦,然后使被保护的伐昔洛韦脱保护,形成伐昔洛韦或其药学上可接受的盐。本发明还涉及盐酸伐昔洛韦的制备及纯化。
[1] [中国发明,中国发明授权] CN201611080343.9 一种手性噁唑啉类NNP型配体及其合成方法和应用
[2] CN200710092592.4伐昔洛韦或其药学上可接受盐的制备
[3] [中国发明,中国发明授权] CN02150810.0 Nα-9-芴甲氧羰基-氨基酸的合成方法
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手
























