嘌呤核苷磷酸化酶简称PNP,是嘌呤补救合成途径的关键酶之一,广泛存在于哺乳动物、寄生虫和微生物中。按照嘌呤核苷磷酸化酶的蛋白结构可分为两类:低分子量的同源三聚体类和高分子量的同源六聚体类。其中哺乳动物和部分微生物(例如鼠伤寒沙门氏菌salmonellatyphimurium,嗜热古菌芝田硫化叶菌Sulfolobus solfataricus等)的嘌呤核苷磷酸化酶属于同源三聚体类,其分子量约为80~100kDa,每个亚基的分子量为30~32kDa,通常此类嘌呤核苷磷酸化酶只能以鸟嘌呤核苷和肌苷作为底物。而同源六聚体类的嘌呤核苷磷酸化酶,底物专一性不强,既可接受鸟嘌呤核苷和肌苷作为底物,也可以腺嘌呤核苷作为底物。其分子量约为150kDa,亚基分子量为25kDa左右.

由于嘌呤核苷磷酸化酶特殊的生物活性,使得其在医药领域得到了较大的应用:①应用于核苷类药物的合成,大量研究表明,核苷类(核苷及其类似物)具有广泛的抗肿瘤抗病毒的效果。②应用于肿瘤靶向治疗;③应用于无机磷和ATPase酶活力等的定量测定.
本发明的目的在于提供一种经过分子改造并具有较高热稳定性的嘌呤核苷磷酸化酶,以及高产量的制备该嘌呤核苷磷酸化酶的方法.
本发明第一方面提供了一种突变的假交替单胞菌属的嘌呤核苷磷酸化酶,与野生型的氨基酸序列相比,所述突变的嘌呤核苷磷酸化酶的氨基酸序列中第98位Asp突变为Tyr.
在另一优选例中,所述突变的嘌呤核苷磷酸化酶具有以下一种或多种特性:
(a)比酶活≥30U/mg,较佳地为≥45U/mg;
(b)储存在碱性环境中;
(c)失活温度≥60℃;
(d)在0-55℃,保存时间≥3h.
在另一优选例中,所述突变的嘌呤核苷磷酸化酶的最适PH值为7.0-9.0,较佳地为7.5-8.5,更佳地为8.0.
本发明第二方面提供了一种分离的多核苷酸,所述多核苷酸编码第一方面所述突变的嘌呤核苷磷酸化酶.
天然核苷是由核糖或脱氧核糖与天然碱基所形成,构成的核糖核苷或脱氧核糖核苷也是种类繁多。天然核苷中的碱基或糖基经过化学修饰形成新的核苷,这种经过人工合成的核苷即为核苷类似物。核苷类似物与核苷酸在化学结构上具有类似性,可以假乱真混入DNA合成的过程中,但因其不具备核苷酸的功能,所以导致合成的核酸链失去了正常的功能。通过氮杂、脱氮、取代等化学修饰方法,对正常碱基的嘧啶和嘌呤进行修饰就可得到核苷类药物。核苷类似物在病毒或肿瘤细胞的遗传物质表达的过程中,被整合到DNA链中,不仅大大降低了聚合酶的活性,也终止了DNA的合成,从而使病毒细胞无法增殖传代,导致病毒的灭亡。因此,核苷类药物在治疗艾滋病毒、乙肝病毒、肿瘤等领域表现出极大的优势。据统计,在目前使用的抗病毒药物中有将近50%是核苷类药物。
核苷类药物中的5-氟尿苷是一种重要的5-氟嘧啶类衍生物,可以制备多种抗癌抗病毒药物,特别是可以做为抗肿瘤新药氟铁龙(DFUR)的前体。2-去氧氟尿苷为脲嘧啶类抗代谢物,属细胞周期特异性抗肿瘤药,可抑制胸腺嘧啶脱氧核苷酸合成酶,阻断脲嘧啶脱氧核苷酸转变成胸腺嘧啶核苷酸,从而影响DNA的合成,起到抗病毒和抗肿瘤的作用。该药对多种肿瘤细胞具有显著的抑制作用,临床上主要用于治疗肝癌、胃肠系癌、乳腺癌和头颈部肿瘤等,与常用抗肿瘤药物5-氟脲嘧啶(5-fluorouracil)相比,具有低细胞毒性和易于在体内被吸收的优点。现有技术中,常采用化学法糖基化得到脱氧氟尿苷,产物中会出现多种同分异构体,并且合成步骤繁多,需要对碱基或核糖基上的活性基团进行修饰保护,且合成过程中大量使用有机溶剂,同时还要使用毒性较大的氟化物、溴化物、重金属等,对环境造成巨大的污染。核苷磷酸化酶可以催化核苷或脱氧核苷的糖苷键的可逆磷酸化反应,提供核糖-1-磷酸,并释放碱基,加入其它碱基可形成另一种新的核苷。因此,核苷磷酸化酶主要用作酶法合成核苷类似物的工具。但是,现有核苷磷酸化酶的转化率较低,制备核苷类药物的成本较高。
2-去氧氟尿苷的制备方法如下:以核苷磷酸化酶PyNP为催化剂,采用不同的核糖供体和碱基受体,制备5-甲基尿苷、5-氟尿苷、5-氮胞苷、5-氟胞苷、胸苷、2-去氧氟尿苷、2’-脱氧-5-氮胞苷、2’-脱氧-5-氟胞苷、2’-脱氧-5-氟尿苷、2’-脱氧-5-氟胞苷、2’-脱氧-5-氟胞苷、2’-脱氧-5-甲基尿苷、阿糖-5-氟尿苷、阿糖-5-氮胞苷和阿糖-5-氟胞苷。在1ml的pH8.0、80mM的磷酸缓冲液中(含有1mM的EDTA),分别加入终浓度为60mM的核糖供体和终浓度为30mM的碱基受体,加入终浓度为1.5U/mL纯化后的核苷磷酸化酶,在50℃、转速为900rpm的恒温振荡反应器中反应。反应3h后,取50μl样品加入到950μl甲醇中以终止反应,利用高效液相色谱HPLC检测产物。检测条件:利用AgilentTC-C18的检测柱,流动相为甲醇:水=95:5,检测波长为254nm,柱温为30℃,流速为1ml/min。根据峰面积计算底物的转化率。其中核糖供体为尿苷、2’-脱氧尿苷、胸苷、2’,3’-双脱氧胸苷或阿糖尿苷。碱基受体为5-甲基脲嘧啶、胸腺嘧啶、5-氟脲嘧啶、5-氮胞嘧啶或5-氟胞嘧啶。
[1] CN201610218675.2一种核苷磷酸化酶、编码基因及其高产菌株和应用
非布司他是一种新型的非嘌呤类黄嘌呤氧化还原酶(XOR)抑制剂,用于治疗慢性高尿酸血症痛风患者。它首先由日本帝人公司于2004年在日本、美国和欧洲申请上市。2008年5月5日,IPSEN公司的非布司他片剂(商品名:ADENURIC)获得了欧盟批准,在法国上市。2009年2月,武田制药公司(Takeda Pharmaceuticals America)的非布司他片(商品名:ULORIC)也获得了美国FDA的批准。
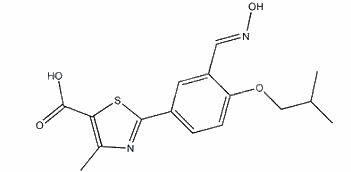
非布司他对XOR具有高度的选择性,并对氧化型和还原型的XOR均有显著的抑制作用。它的抑制XOR的Ki和Ki'值分别为0.6和3.1nM。而非布司他在高达100μM的浓度下,对涉及体内嘌呤和吡啶代谢的以下酶活性没有影响:鸟嘌呤脱氨基酶、次黄嘌呤-鸟嘌呤磷酸核糖转移酶、嘌呤核苷磷酸化酶、芳香磷酸核糖转移酶以及乳清酸核苷酸脱羧酶等。并且非布司他对XOR的抑制作用不受酶的氧化还原状态的影响。非布司他是一种特异性的新型XOR抑制剂,其作用强于别嘌醇。它主要通过肝脏代谢,不依赖肾脏排出,因此对轻中度肾功能不全者也是安全有效的。非布司他杂质7是非布司他合成过程中产生的杂质。
非布司他杂质7的制备方法如下:首先将2-[3-((羟基亚氨基)甲基)-4-(2-甲基丙氧基)苯基]-4-甲基噻唑-5-羧酸乙酯(100克)的甲醇(500毫升)溶液加入预先冷却的氢氧化钠中。将反应混合物加热至75℃并在相同温度下搅拌30分钟。然后向反应混合物中加入水(5克)并将其冷却至25℃。接下来,向反应混合物中加入甲醇(100ml)和水(100ml),使用盐酸水溶液将pH调节至2.0-3.0,并继续搅拌反应混合物5小时。
过滤沉淀的固体并用甲醇和水洗涤,然后干燥所得材料。将所得物质中加入四氢呋喃(270ml)和碳(2.25g),搅拌反应混合物30分钟并通过hyflow过滤。从滤液中完全蒸馏出溶剂。将所得化合物中加入甲醇(450ml),在60℃下搅拌30分钟。将反应混合物冷却至25℃并继续搅拌2小时。最后,过滤沉淀的固体并干燥,得到纯的非布司他杂质7。产量为70克。
[1] WO2011141933 PROCESS FOR THE PREPARATION OF 2-[3-CYANO-4-(2-METHYLPROPOXY)PHENYL]-4-METHYLTHIAZOLE-5-CARBOXYLIC ACID AND ITS PHARMACEUTICALLY ACCEPTABLE SALTS
黄素腺嘌呤二核苷酸(FAD)是一种黄素蛋白的氧化还原辅因子,它在生物体内起着重要的作用。FAD参与电子从底物到电子接受体的传递,是黄素酶类的辅酶。它广泛分布在各个行业,如银行、计算机、教育、金融、政府和卫生等。
除了FAD,黄素腺嘌呤二核苷酸可能还有其他首字母缩略词。如果您想了解黄素腺嘌呤二核苷酸的英文版本,请向下滚动到底部。
黄素腺嘌呤二核苷酸在生物体内具有吸收带,在450、375和260纳米处有吸收峰。然而,一旦与蛋白质结合,吸收带的状态会改变。
黄素腺嘌呤二核苷酸在好氧生物和厌氧生物的体内广泛分布。它还参与丙酮酸脱氢酶复合物的组成,以及一氧化氮的合成。此外,黄素腺嘌呤二核苷酸还是一种电子载体,包括琥珀酸脱氢酶、αc-酮发二酸脱氢酶、凋亡诱导因子2、叶酸/FAD依赖的tRNA甲基转移酶和N-羟基化黄素蛋白单加氧酶的辅因子。
黄素腺嘌呤二核苷酸可以通过焦磷酸化酶合成,该酶将FMN和ATP转化为FAD。
核苷是由结合于核糖或脱氧核糖的碱基如嘌呤或嘧啶组成的糖基胺。核苷类似物被广泛用作抗病毒剂和抗癌剂,因为它们在RNA或DNA合成中能够用作反转录酶抑制剂或链终止剂。核苷磷酸化酶是广泛分布于哺乳动物细胞和细菌中并在核苷代谢补救途径中起关键作用的转移酶。核苷的制备方法如下:
6-巯基嘌呤核苷如前述进行制备。将1ml转糖基活性约为12个单位/ml细胞裂解物的催化剂(细胞裂解物)加入至150ml溶液中,所述溶液在80℃下-5mM6-巯基嘌呤,和-30mM磷酸钾缓冲液,pH7。该测定使用上文所述的条件在作为共溶剂的10%(v/v)DMSO中进行。在80℃下5小时后,应用具有3000-Da截留值的Amiconultra-4CentrifugalFilterDevices(Millipore,Bedford,MA)通过在4℃以2000×g离心30分钟将所述反应混合物过滤,并回收滤液。所述反应的生物转化产率高于50%。将得到的6-巯基嘌呤核苷(6-巯基嘌呤核糖核苷和6-巯基嘌呤脱氧核糖核苷)通过HPLC进行分析。
[1] (CN102770532) 用于核苷合成的热稳定生物催化剂结合物
鸟苷或2′-脱氧鸟苷在工业上主要是通过提取/分离DNA(脱氧核糖核酸)或RNA(核糖核酸)的水解产物来生产,因为化学合成鸟苷或2′-脱氧鸟苷的产率极低。然而,DNA水解产物中除了含有所需的2′-脱氧鸟苷之外,还含有2′-脱氧腺苷、2′-脱氧胞苷和胸苷。RNA的水解产物中除了所需的鸟苷之外,还含有腺苷、胞苷和尿苷。因而,为了只收取鸟苷或2′-脱氧鸟苷,必须实施复杂的提取/分离过程,因此不可避免地提高了产品的成本。

另一方面,业已报道了一种方法,在该方法中,使作为原料的核苷(或脱氧核苷)和核酸的碱基(nucleic acid base)在核苷磷酸化酶的作用下进行碱基交换反应,由此获得所要的核苷(或所要的脱氧核苷)(Hori,N.,Watanabe,M.,Yamazaki,Y.,Mikami,Y.,Agric.Biol.Chem.,53,197-202(1989))。通过这种方法,可以容易地制备腺苷和2′-脱氧腺苷(日本专利申请说明书No.11-46790“制备嘌呤核苷化合物的方法”)。为了用这种酶合成反应方法得到鸟苷和2′-脱氧鸟苷,至少在反应所需的酶能发挥作用的pH范围内,鸟嘌呤必须是溶于水的。一般认为,为了使得酶反应顺利进行,最好反应底物的溶解度至少等于酶的米氏常数(Km)或更高。但是,由于鸟嘌呤的水溶解度不超过几个ppm,所以上述酶合成反应方法不能实际采用。鉴于这种陷于僵局般的状况,需要开发一种制备鸟苷或2′-脱氧鸟苷的有效方法。
本文介绍了合成和纯化2'-氟-2'-脱氧尿苷的方法,这是一种重要的有机化合物,具有广泛的应用领域。
简述:2'-氟-2'-脱氧尿苷,英文名为2'-Fluoro-2'-deoxyuridine,分子式为C9H11FN2O5,可溶于甲醇,熔点为147.0- 153.0℃。2'-氟-2'-脱氧尿苷为核苷酸类衍生物,常用作用作医药中间体。
1. 合成:
步骤(1)将尿苷、碳酸二苯酯加于二甲基甲酰胺中,升温溶解,溶液澄清后,保持温度条件下加入催化剂碳酸氢钠,第二次升温反应,反应完成后,自然降温至室温,搅拌,过滤,滤饼烘干得化合物2;
步骤(2)将化合物2、无水氟化钾、催化剂三氟化硼乙醚溶于溶剂中,升温反应,反应完成后过滤浓缩,得化合物3,即2’?氟?2’?脱氧尿苷。
其中,步骤(1))中化合物1:碳酸二苯酯的摩尔比为1:1.1,升温溶解的温度为58~62℃,第二次升温温度为78~82℃,室温为20~30℃,搅拌时间为1~3小时。步骤(2)中溶剂为二甲基甲酰胺或氯苯,化合物2:无水氟化钾的重量比为1~2:1,升温反应温度为120℃,反应时间为10-14小时。
2. 纯化:
(1)将粗品2’-氟-2’-脱氧尿苷溶于乙酸乙酯,加入三乙胺,控温下滴加酰化试剂反应;
(2)反应完成后加入甲醇,搅拌,降温析晶,过滤,滤饼用乙酸乙酯洗涤,滤饼烘干的化合物2;
(3)化合物2溶解于甲醇中,加入氢氧化钠,反应完全后过滤,滤饼烘干得2’-氟-2’-脱氧尿苷。
实验中,酰化试剂为醋酐或苯甲酰氯当中的一种。当步骤(1)中加入的酰化试剂为醋酐时,需要加入催化剂,催化剂为4-二甲氨基吡啶,2’-氟-2’-脱氧尿苷:三乙胺:酰化试剂的重量比为1:1~3:1~2。 步骤(1)控制反应温度在0-20℃,步骤(2)降温温度为-20~10℃,步骤(3)化合物2与氢氧化钠的摩尔比为1:1。
3. 应用:合成2’-氟-2’-脱氧腺苷
选取胸苷磷酸化酶(Thymidinephosphorylase,TP)与嘌呤核苷磷酸化酶(Purinenucleosidephosphorylase,PNP)作为生物催化剂,催化 2’-氟-2’-脱氧尿苷与腺嘌呤进行转糖基化反应生成2’-氟-2’-脱氧腺苷。具体步骤如下:
(1)TP与MF13的酶液制备;取TP湿菌体0.5g,加入10mL100mM磷酸缓冲液 (pH=7.0)重悬,设定超声破胞仪的功率为 400W,超声3s间歇6s,破碎细胞20min,将破胞后的菌液以13000rpm 离心20min,取上清液保存于-20℃冰箱备用。
(2)2'-氟-2'-脱氧尿苷3.125g,腺嘌呤4.175g,TP酶液100mL,MF13 酶液100mL,加入100mM磷酸缓冲液 (pH=7.0)300mL,反应温度48℃,转速800r/min,反应期间每隔12h取一次样,将样品在 100℃水浴 12s灭活,使用高效液相色谱仪检测。
参考文献:
[1] 滕海东. 2’--氟--2’--脱氧腺苷的核苷磷酸化酶催化合成技术研究[D]. 浙江:浙江大学,2020.
[2] 平江县吉成科技有限责任公司. 一种2’-氟-2’-脱氧尿苷的合成方法:CN202011452956.7[P]. 2021-03-16.
[3] 平江县吉成科技有限责任公司. 一种2’-氟-2’-脱氧尿苷的纯化方法:CN202011443647.3[P]. 2021-02-26.
近期关于奈拉滨的合成研究不断涌现,为其应用领域的拓展和开发提供了新的机遇。
简介:奈拉滨(nelarabine,1)的商品名:Arranon,化学名:2-氨基-9-β-D-阿糖呋喃糖腺嘌呤-6-甲氧基-9H-嘌呤。是细胞毒素脱氧鸟苷ara-G的水溶性前药,用于治疗对至少两种化疗方案无反应或治疗后又复发的急性T细胞淋巴母细胞性白血病(T-ALL)和T-细胞淋巴母细胞性淋巴瘤(T LBL)。其进入血液后能在腺苷脱氨酶的作用下迅速脱甲基,转化成ara-G(9-β-D-阿糖呋喃糖鸟嘌呤)的三磷酸盐,通过抑制DNA的合成,诱导易感细胞脱噬而起作用。
合成:
1.传统合成方法:①酶促法以6-甲氧基-2-氨基嘌呤和阿糖尿苷为原料,在生 物酶嘌呤核苷磷酸化酶和尿嘧啶核苷磷酸化酶催化下,经过碱基转移反应,得到1,总收率15%,转化酶价格昂贵,菌种难以得到,产品的提取采用离子交换树脂,难以工业化。②以6-氯鸟苷为原料,经6-位甲氧基化、酰化、选择性脱去2'-位乙酰基,磺酰化、乙酰化构型转化、脱保护基等6步反应,合成1,反应路线长,总收率8%。③用6-甲氧基鸟嘌呤和氯代阿拉伯糖在重金属催化剂作用下缩合,进而脱保护基得到1,方法中重金属催化剂有毒有害。综上可以看出,目前对奈拉滨的研究比较少,而且不管是化学法还是生物法得到的奈拉滨的收率都不高。并且也没有实现工业化。随着奈拉滨需求量的日益增加,其新的合成方法的研究势在必行。
2.有研究以细胞毒素阿糖鸟苷(2)为原料,经过乙酰化保护制得 2',3',5'-三-O-乙酰基阿糖鸟苷(3),3经三氯氧磷氯代制得6-氯-2',3',5'-三-O-乙酰基阿糖鸟苷(4), 4再经甲氧基化反应制得1,总收率73%。

2.1 2',3',5'-三-O-乙酰基阿糖鸟苷(3)
将2(10.0 g,35.30 mmol)加入250 ml三口瓶中,加入乙酐(100.0 ml,1.10 mol)和4-二甲胺基吡啶(DMAP, 0.2 g,1.80 mmol),加热至回流反应3 h。减压蒸除过量的乙酐,加入无水乙醇(10 ml),减压蒸除,得淡黄色油状物,再加入无水乙醇(50 ml),加热溶解,将溶液转移到烧杯中,加入5%活性炭(0.5 g) 脱色,趁热过滤,滤液静置析晶2 h,过滤,干燥,得白色固体3(13.4 g,93.0%),mp :212~214 ℃,纯度>98.0%。
2.2 6-氯-2',3',5'-三-O-乙酰基阿糖鸟苷(4)
将3(10.0 g,24.40 mmol)加入二氯乙烷(100 ml) 中,加入三氯氧磷(11.0 ml,122.00 mmol),回流反应5 h,倒入冰水(500 ml)中,充分搅拌,分液,有机相用饱和碳酸氢钠溶液(50 ml×3)洗至pH 7,加入活性炭脱色,过滤,滤液用无水硫酸钠干燥后过滤,滤液减压蒸除溶剂,得淡黄色油状物,加入无水乙醇(30 ml),加热溶解,趁热过滤,滤液静置,析出白色固体,过滤,干燥,得白色固体4(9.1 g, 87.0%),mp:194~196 ℃。纯度>98.0%。
2.3 奈拉滨(1)
将4(10.0 g,23.40 mmol)溶于甲醇(100 ml) 中,加入碳酸钠(3.7 g,35.10 mmol),回流反应5 h,TLC[展开剂:甲醇∶二氯甲烷(3∶7),Rf =0.45] 显示反应完全,冷却至室温,过滤除去过量的盐,母液减压蒸除溶剂,得淡黄色油状物,加入去离子水(20 ml),加热溶解,静置析晶,过滤,干燥,得到白色固体1(6.3 g,90.0%),mp:202~204 ℃。
2.4 该方法中原料 2可从价廉易得的鸟苷大量合成得到,使用的其他试剂均为常用的化工原料,产率较高,适合工业化生产。
参考文献:
[1]夏然,孙莉萍,杨西宁等.奈拉滨的合成[J].中国医药工业杂志,2015,46(12):1278-1280.DOI:10.16522/j.cnki.cjph.2015.12.003.
[2]梁平,尹先清,李卫佳.奈拉滨的合成研究进展[J].精细化工中间体,2008(05):8-10.DOI:10.19342/j.cnki.issn.1009-9212.2008.05.003.
利巴韦林是一种广谱抗病毒药物,其化学名为Ι-β-D-呋喃核糖基-1H_1,2,4-三氮唑-3-羧酰胺。它通过抑制单硝酸、次黄嘌呤核苷脱氢酶的活性,干扰DNA的合成,阻止病毒复制,对多种DNA病毒和RNA病毒均有抑制作用。利巴韦林无交叉耐药性,无诱导干扰素作用,但具有免疫抑制作用。
利巴韦林于1972年由Witkowski等合成,1986年获得美国FDA批准上市,主要用于气雾罩治疗婴幼儿合胞病毒感染。1998年7月,其适应证得到扩大。

利巴韦林的生产可以采用发酵法、酶促法和化学法。
发酵法的缺点是需要每次培养菌体,培养时间长,生产效率低,产生多种副产物,纯化困难。
酶促法需要较难获得的核糖磷酸和昂贵的纯酶。利巴韦林的产率受到核苷磷酸化酶活性的限制,以及以肌苷作核糖供体时产率低于85%。
化学合成是一种常用的利巴韦林制备方法。例如,可以通过在磷酸二(对硝基苯基)酯存在下,将3-甲酯基三唑和四乙酰基核糖的1:1混合物在160-165°C下熔融来制备利巴韦林。然而,这种方法的产率低于理论值,存在大量副产物,成本较高。
一种优化的利巴韦林制备方法包括以下步骤:
A、以肌苷为原料,加入酸和催化剂I进行酰化反应,生成四乙酰核糖;
B、将步骤A的四乙酰核糖和1,2,4-三氮唑-3-羧酸甲酯分别用活性炭处理后混合,然后加入催化剂II进行缩合反应得到缩合物;
C、将步骤B的缩合物在氨和甲醇中氨解生成利巴韦林粗品;
D、将步骤C的利巴韦林粗品进行精制处理得到利巴韦林纯品。
在步骤A中,催化剂I是NaSiO3与Na2HPO4的混合物,酸可以是醋酐、醋酸中的一种或两种。步骤B中的催化剂II是经过活性炭处理的磷钨钼杂多酸,然后用氨水对其进行表面处理。
本文将介绍2’-脱氧鸟苷的合成方法,这对于理解该化合物的制备过程以及在药物合成和化学领域的应用具有重要意义。
背景:近年来,人们对核苷及其衍生物在抗病毒和抗肿瘤方面的作用越来越感兴趣。1992年,C. Perigaud等人对核苷类抗病毒药物进行了综述。2′-脱氧鸟苷作为合成寡脱氧核苷酸等多种抗病毒、抗肿瘤核酸药物的重要原料,在市场上需求广泛。然而,脱氧鸟苷的化学合成一直存在困难,迄今为止仍未有成熟的方法被报道。目前,获得脱氧鸟苷的主要方法是采用DNA作为原料,首先经核酸酶水解生成脱氧核苷酸,然后再通过磷酸单酯酶水解形成脱氧核苷。这种方法成本高,而且DNA来源有限,限制了其大规模生产的可能性。
关于酶法合成脱氧鸟苷的报道,日本横关键三等应用肺炎克雷伯氏菌(Klebsiella pneumoniae)生成脱氧鸟苷的方法,但转化率仅为2 8%。Yokozeki等使用产气肠杆菌(Enterobacter aerogenes)为酶源,以2′-脱氧尿苷和鸟苷为底物,2′-脱氧鸟苷转化率仅为18%。其它亦有用胸苷和2,6-二氨基嘌呤生成2,6-二氨基2′-脱氧核苷,再用腺苷脱氨酶处理得到脱氧鸟苷的方法,但工艺步骤多,成本较高。
合成:
1. 方法一:
以脱氧胸腺嘧啶核苷(Thymidine, dTR)为脱氧核糖的供体,鸟苷酸(Guanosine monophosphate, GMP)为碱基鸟嘌呤的供体,在核苷磷酸化酶的作用下合成脱氧鸟苷。具体步骤如下:
(1)乙酰短杆菌的培养和湿菌体制备
在1L的带挡板的锥形瓶中装入发酵培养基200mL,于121℃灭菌20min,冷却后从新鲜、长势良好的乙酰短杆菌斜面上接种,36℃培养16 h,然后将培养液10000r/min离心10min,弃去上清液,所得菌体用30mmol/L磷酸盐缓冲液(pH7.0)洗涤2~3次,作为酶源冷冻备用。
(2)脱氧鸟苷酶法合成及工艺优化
20mmol/L的脱氧胸腺嘧啶核苷(dTR),20mmol/L的鸟苷酸(GMP),5%的湿菌体,100mL 30mmol/L pH6.98磷酸盐缓冲液作为标准反应混合物,在60℃的恒温水浴槽中搅拌反应,反应时间为6h。在不改变其它条件的情况下,依次考察不同的反应温度、菌体量、反应pH、磷酸盐缓冲液浓度、底物浓度、底物比率和反应时间对酶反应的影响,优化其反应条件。
(3)酶反应液中脱氧鸟苷的分离纯化
取适量的脱氧鸟苷反应液,高温灭活后用硅藻土过滤,在其中加入5mmol/L的四硼酸钠,再通过pH中性的氯型阴离子树脂进行分离,以0.5倍柱体积每小时的流速上样洗脱,上样结束后用0~0.2mol/L的NaCl进行梯度洗脱,分布收集洗脱液。取收集到的脱氧鸟苷洗脱液脱盐浓缩结晶,得到脱氧鸟苷样品。
2. 方法二:
以β-胸苷和鸟嘌呤为原料,采用发酵乳杆菌菌体浆状物为生物催化剂,在聚乙二醇10000/磷酸氢二钾双水相系统下进行生物催化反应,得到2’-脱氧鸟苷产物。在最优化条件下,底物β-胸苷加入量达到16.5g/L,胸苷转化率达89%,产物2’-脱氧鸟苷达16.1g/L。
3. 方法三:
(1)培养乙酰短杆菌(Brevibacterium-acetylicum)QD96-CGMCCNo.0472;(2)然后将步骤(1)获得的菌体加入底物溶液,其底物溶液中,含有脱氧核糖受体、脱氧核糖供体和磷酸缓冲液,反应,然后从反应产物中收集2’-脱氧鸟苷。该方法使用乙酰短杆菌QD96的酶合成2’-脱氧鸟苷,可低成本,高收率地获得2’-脱氧鸟苷,其转化率可达60%以上。
参考文献:
[1]李喻,窦洁,曹静,等. 乙酰短杆菌酶法合成2'-脱氧鸟苷[J]. 药物生物技术,2011,18(2):119-123.
[2]乐山市瑞和祥生物制药有限公司. 一种核苷类药物中间体2’-脱氧鸟苷的生产方法:CN201510140553.1[P]. 2015-08-12.
[3]南通秋之友生物科技有限公司,江苏秋之友核酸药物工程技术研究中心有限公司. 用乙酰短杆菌的核苷磷酸化酶合成2’-脱氧鸟苷的方法:CN201010611733.0[P]. 2011-09-07.
[4]韩国威. 单线态氧损伤2'-脱氧鸟苷和8-氧代脱氧鸟苷机理的理论研究[D]. 郑州大学, 2012.
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手

























