异丁基氯化镁是一种活泼的试剂,可以与具有活泼氢的化合物发生反应。这些化合物包括H2O、ROH、RC≡CH…、醛、酮、酯、酰卤、腈、环氧乙烷、卤代烷、二氧化碳、三氯化磷、三氯化硼、四氯化硅等。
异丁基氯化镁可以用于制备普瑞巴林,一种抗癫痫药物。制备普瑞巴林的方法包括与格氏试剂异丁基氯化镁的加成反应、脱保护、开环和手性拆分等步骤。

制备普瑞巴林的第一步反应操作如下:
在500mL干燥的三颈烧瓶中加入溴化亚铜-二甲硫醚(0.41g,2mmol),无水四氢呋喃50mL。将温度降至-20℃,在30分钟内滴加新制的化合物3(异丁基氯化镁40mmol)四氢呋喃溶液20mL。将温度降至-40℃,在30分钟内滴加化合物2(4.68g,40mmol)的四氢呋喃溶液40mL,保持搅拌过夜,在-40℃至-20℃范围内。加入饱和氯化铵溶液淬灭反应,整个实验过程在N2保护下进行。将温度升至室温,分液,用乙酸乙酯萃取水层3次。合并有机层,用饱和食盐水洗涤2次,加入无水硫酸镁干燥,用旋转蒸发仪除去溶剂得到化合物4,收率为80%,无需纯化即可用于下一步反应。
[1]FromJournalofOrganicChemistry,65(7),2231-2235;2000
[2][中国发明]CN201711177781.1一种抗癫痫药普瑞巴林的制备方法
4-氯-2-氟苯甲醛是一类重要的医药、农药及染料的中间体,特别是合成吲唑类、喹唑啉类的原料。例如拜耳医药股份公司的专利CN109153665A公开了利用4-氯-2-氟苯甲醛为原料制备4-氯-2-氟-5-硝基苯甲醛,然后制备2-取代的吲唑化合物。
一种4-氯-2-氟苯甲醛的合成方法,其化学反应式为:

所述方法包括如下步骤:
(1)4-氯-2-氟溴苯与金属镁或格氏试剂反应制成格氏液;
(2)向步骤(1)的格氏液中滴加DMF,反应完毕后,用稀盐酸水解,得到4-氯-2-氟苯甲醛。
根据本发明的一个实施方式,所述格氏试剂选自乙基氯化镁、乙基溴化镁、丙基氯化镁、丙基溴化镁、异丙基氯化镁、异丙基溴化镁、丁基氯化镁、丁基溴化镁、环己基氯化镁、环己基溴化镁、甲基氯化镁、甲基溴化镁中的任意一种。
根据本发明的一个实施方式,步骤(1)中4-氯-2-氟溴苯与金属镁或格氏试剂的摩尔比为1:1-3,优选1:1-1.6;
步骤(2)中4-氯-2-氟溴苯与DMF的摩尔比为1:1-4,优选1:1-2。
根据本发明的一个实施方式,所述稀盐酸的浓度不作具体限制,通常为5%-25%,优选的浓度为8-15%。
根据本发明的一个实施方式,步骤(1)格氏反应的温度为-15℃-70℃,优选20-70℃;本发明出于节约能源考虑,格氏反应温度优选20-40℃;
步骤(2)滴加DMF的反应温度为-20℃-60℃,优选-15℃-40℃;本发明出于节约能源考虑,滴加DMF的反应温度优选20-40℃。
根据本发明的一个实施方式,步骤(1)格氏反应的时间为1-12h,优选2-4h。
步骤(2)滴加DMF的反应时间为1-12h,优选5-7h。
本文将介绍合成2-溴-5-乙酰基吡啶的具体步骤和操作技巧,通过深入探讨合成过程中的关键因素,旨在为读者提供合成2-溴-5-乙酰基吡啶的指导和参考。
背景:2-溴-5-乙酰基吡啶是一种重要的药物中间体,用于修饰抗癌药物酪氨酸激酶抑制剂伊马替尼的嘧啶环。目前,已报道的2-溴-5-乙酰基吡啶的制备方法为2,5?二溴吡啶与正丁基锂/四氢呋喃在?78℃反应。然而,使用的正丁基锂危险性大,操作性差,对合成人员和工作环境要求高,因此仅适用于样品制备不适合工业化生产。另外,也有报道使用异丙基氯化镁四氢呋喃溶液作为金属化试剂的方法。但是,由于异丙基氯化镁四氢呋喃溶液浓度低,导致反应缓慢且原料无法完全消耗。
制备:可使用2,5?二溴吡啶合成2-溴-5-乙酰基吡啶,具体步骤如下:在步骤S1中,以2,5?二溴吡啶为底物,滴加异丙基氯化镁的2?甲基四氢呋喃溶液,生成4?溴吡啶氯化镁;接着,在步骤S2中,向步骤S1的反应液中继续滴加乙酸酐,使其与4?溴吡啶氯化镁发生反应,生成5?乙酰基?2?溴吡啶。该制备方法可降低原料危险性,提高实验安全性和操作性,并且具有较高的实验重复性。同时,减少了三废的产生,降低了环保压力;使用异丙基氯化镁的2?甲基四氢呋喃溶液可以提高反应速度和收率,并且能够得到纯度较高的产品。
其中异丙基氯化镁的2?甲基四氢呋喃溶液摩尔浓度为2.5?4mol/L。在步骤S1中,2,5?二溴吡啶与异丙基氯化镁的摩尔比为1:(1?1.2)。滴加异丙基氯化镁的2?甲基四氢呋喃溶液温度保持在5?10℃,滴加结束后继续反应0.5?2小时。2,5?二溴吡啶与乙酸酐的摩尔比为1:(1?1.2)。在步骤S2中,滴加乙酸酐的温度为5?10℃,滴加结束后继续反应0.5?2小时。接着,在步骤S3中,反应结束后,将反应液滴加到柠檬酸水溶液中并分层,然后用乙酸乙酯进行萃取。合并有机相后进行水洗、干燥、过滤和浓缩,得到粗品。最后,在步骤S4中,粗品用石油醚/乙酸乙酯2:1打浆,得到纯化的2-溴-5-乙酰基吡啶。
参考文献:
[1] 苏州昊帆生物股份有限公司. 一种5-乙酰基-2-溴吡啶的制备方法:CN202210674864.6[P]. 2022-09-06.
[2] 朱菊,李帅,李馨阳,等. 甲磺酸伊马替尼的合成新方法[J]. 中国药物化学杂志,2020,30(1):26-29. DOI:10.14142/j.cnki.cn21-1313/r.2020.01.004.
环己基氯化镁,英文名:chlorocyclohexylmagnesium,CAS号:931-51-1,分子量:142.91000,密度:0.871 g/mL at 25oC,沸点:80.7oC at 760 mmHg,分子式:C6H11ClMg,闪点:-40°F,为透明非常暗棕色溶液,密封贮藏,储存在阴凉、干燥的地方,远离火源,易燃易爆区,腐蚀区,水区。避免阳光直射。室温储存。常用氮气保护。

将三甲氧基苯(1.0062g,6mmol)溶液加入20mL干燥的四氢呋喃中,在N2下滴入正丁基锂(2.6mL,2.5M正己烷溶液,6.6mmol)。室温搅拌4.5小时,冷却至-78℃,滴加二环己基膦。混合物搅拌30分钟后,将反应系统加热至室温,再搅拌18小时。乙酸乙酯萃取,水洗,无水硫酸钠干燥,柱层析纯化(石油醚/乙酸乙酯=10-15/1),所得环己氯化镁产品0.5514g。收率是25%。1HNMR(300MHz,CDCl3)δ-6.08(d,J=1.5Hz,2H),3.81(s,3H),3.77(s,6H),2.30-2.16(m,2H),1.92-1.54(m,8H),1.48-0.90(m,12H);31PNMR(121 MHz,CDCl3)δ-14.6[1]。
1、在专利CN201610030857.7实施例十六中介绍了[Fe(PR3)X3][(R1NCHnCHnNR1)CH](R1为2,6-二异丙基苯基,R为环己基,n为1,X为Br)催化的环己基氯化镁与磷酸二乙-2-吡啶酯的交叉偶联反应:在经过脱水脱氧处理过的反应瓶中,在氩气保护下依次加入催化剂的四氢呋喃溶液1.0毫升(5.0×105毫摩尔/毫升),磷酸二乙-2-吡啶酯0.12克(0.5毫摩尔)和四氢呋喃2.0毫升。在0°C下缓慢滴加环己基氯化镁的四氢呋喃溶液2.0毫升(1.0毫摩尔/毫升),搅拌5分钟。升至25℃搅拌反应10小时,用去离子水终止反应,反应产物用乙酸乙酯萃取,层析柱提纯(以石油醚为展开剂),产率80%[2]。
2、专利CN200780032144.3实施例1中步骤B关于[4-(环己基-羟基-甲基)-苯基1-(4-异丙基-哌嗪-1-基)-甲酮的合成,在-78°C向4-(4-异丙基-哌嗪-1-羰基)-苯甲醛(351mg,1.35mmol)在THF(15mL)内的溶液中加入环己基氯化镁(2.0M在Et2O中的溶液;0.81mL,1.62mmol)。让该混合物温热至室温并且搅拌5小时。用饱和NH4Cl水溶液中止该反应,倒入H2O内,并且用3批CH2Cl2萃取。将合并的有机层干燥(Na2SO4)并浓缩。通过制备反相HPLC来纯化残余粗产物,获得了目标产物[3]。
[1]ZHEJIANG UNIVERSITY. Structure and method for synthesizing and using dialkyl(2,4,6- or 2,6-alkoxyphenyl)phosphine and its tetrafluoroborate:US200913383208[P]. 2015-04-14.
[2]苏州大学. 一种离子型铁(II)配合物及其制备方法与应用:CN201610030857.7[P]. 2016-05-04.
[3]詹森药业有限公司. 组胺H<sub>3</sub>受体的取代的苯甲酰胺调节剂:CN200780032144.3[P]. 2009-08-19.
3-巯基丙酸甲酯是一类重要的化工原料,可用于制备异噻唑啉酮和4-氯-7-甲基噻吩并[3,2-D]嘧啶等化学品。此外,它还是合成降血脂药物辛伐他汀侧DMB-S-MMP3-[(2,2-二甲基-1-氧代丁基)硫]丙酸甲酯的原料。

CN115197109B公开了一种制备3-巯基丙酸甲酯的方法,使用改性树脂装填反应管,使液体混合物从上至下流过装填有改性树脂的反应管,经过改性树脂催化后发生加成反应,得到3-巯基丙酸甲酯。
液体混合物包括摩尔比为1:(1-5)的丙烯酸甲酯与硫化氢。
改性树脂的制备方法简单,通过氯化镁、多聚甲醛和三乙胺进行树脂改性,反应条件温和,催化剂制备成本低。
3-巯基丙酸甲酯为可燃液体,吞咽会中毒,吸入致命,皮肤接触有害。对水生生物毒性极大,具有长期持续影响。
2-二吡啶基酮是一种常温常压下为白色或浅黄色固体粉末的化合物,属于吡啶类衍生物,可用作有机合成与医药化学的中间体,以及农药分子和有机催化反应中含氮配体的合成。

图1 展示了2-二吡啶基酮的合成路线。
在搅拌的情况下,将正丁基锂的正己烷溶液慢慢滴加到溶解于四氢呋喃中的2-溴吡啶中,反应混合物在低温下搅拌1小时后,再加入氯甲酸乙酯。随后将反应混合物升至室温,在室温下继续搅拌反应12小时。反应结束后,加入水淬灭反应,然后用二氯甲烷萃取水层并用NaHCO3和盐水洗涤合并的有机物,最后用MgSO4干燥,对溶液进行过滤,减压浓缩得到目标产物。

图2 展示了另一种2-二吡啶基酮的合成路线。
在氮气气氛且室温下将吡啶甲酸和无水四氢呋喃加入反应容器中,在冰浴中冷却反应。然后慢慢加入异丙基溴化镁,再加入2-碘吡啶和异丙基氯化镁-氯化锂配合物,反应混合物在低温下搅拌2小时。反应结束后,用二氯甲烷萃取水层三次,然后将有机层合并,用无水MgSO4干燥,减压浓缩得到目标产物。
2-二吡啶基酮主要用作有机合成与医药化学中间体,可用于农药分子和催化反应中含氮配体的合成。在有机合成转化中,2-二吡啶基酮中的羰基基团可以在还原的条件下转变为羟基基团,也可以和羟胺反应生成相应的肟类产物。
[1] Gass, Ian A. et al Inorganic Chemistry, 50(7), 3052-3064; 2011
[2] Demkiw, Krystyna et al Journal of Organic Chemistry, 81(8), 3447-3456; 2016
2,4,6-三氟苯甲酸(2,4,6-Trifluorobenzoic acid)是合成拉司米地坦的关键中间体,拉司米地坦是中枢神经系统渗透性、选择性的5-羟色胺1F亚型(5-HT1F)口服激动药,于2019年10月获得美国FDA批准,是20多年来FDA批准的首个用于成人急性偏头痛治疗药。

2,4,6-三氟苯甲酸
目前,合成2,4,6-三氟苯甲酸时,主要以三氯苯甲酰氯、五氯苯腈或三氟苯为原料;其中,采用三氯苯甲酰氯为原料时,通过氟代反应和水解反应合成,通过该方法制备的摩尔收率低,制备成本高;采用五氯苯腈为原料时,通过依次对原料氟化、脱氯、水解制备得到产品,但该过程需要使用大量强酸和强碱,会产生有毒和腐蚀性强的废液,操作危险性高且造成了环境污染,规模化生产受限;采用三氟苯为原料时,将三氟苯滴加到丁基锂的正己烷溶液中,再加入固体二氧化碳,最后利用碱提取产品,该过程使用的丁基锂具有危险性,且反应需要在无水、无氧和低温条件下进行,对反应设备要求高,难以实现工业化生产。
2,4,6-三氟苯甲酸的合成分为三步:1)使1,3,5-三氯苯氟化,得到1,3,5-三氟苯;2)使1,3,5-三氟苯发生溴化反应,得到2,4,6-三氟溴苯;3)使2,4,6-三氟溴苯发生格氏反应,得到产品[1]。
1)在氮气保护下,向反应瓶中加入18.15g 1,3,5-三氯苯、54.50g N,N-二甲基甲酰胺、18.6g氟化钾、0.32g四丁基溴化铵进行氟化反应,氟化反应温度为150~160℃,当GC检测1,3,5-三氯苯的含量小于0.2%时,反应完毕,得到了氟化反应体系;其中,1,3,5-三氯苯、N,N-二甲基甲酰胺、氟化钾、四丁基溴化铵的摩尔比为1:7.46:3.20:0.01;对氟化反应体系进行减压蒸馏处理,对所得馏分进行减压精馏处理,得到12.53g第一产物1,3,5-三氟苯的GC纯度为99.3%,摩尔收率为94.9%。
2)向三口瓶中加入12.53g 1,3,5-三氟苯、25mL二氯甲烷、0.50g三氯化铁;将15.98g溴素溶于13mL二氯甲烷中,形成溴素溶液,溴素与二氯甲烷的质量体积比为1g:0.81mL;在30℃下向三口瓶中滴加溴素溶液发生溴化反应,滴加速率为1.66g/min,当GC检测中间体1,3,5-三氟苯的含量小于0.2%,反应结束,得到了溴化反应体系;其中,1,3,5-三氟苯、二氯甲烷、三氯化铁、溴素的摩尔比为1:6.25:0.03:1.05;对溴化反应体系依次碱液洗涤处理、水洗涤处理、对水洗涤处理得到的有机相进行减压蒸馏,得到19.66g第二产物,其结构为2,4,6-三氟溴苯,2,4,6-三氟溴苯,GC纯度为99.2%,摩尔收率为98.2%。
3)在氮气保护下,向三口瓶中加入50mL 2mol/L异丙基氯化镁的四氢呋喃溶液(0.1mol),0℃下滴加19.66g 2,4,6-三氟溴苯,滴加速率为0.98g/min,控制滴加过程中体系温度为0-10℃;其中,2,4,6-三氟溴苯与异丙基氯化镁的摩尔比为1:1.07;在0-10℃下,向体系中通入干燥的二氧化碳气体,当HPLC检测2,4,6-三氟溴苯的含量小于0.2%时,反应结束;在0-10℃下,向格氏反应体系中滴加10%盐酸,用50ml甲基叔丁基醚萃取格氏反应体系,通过旋蒸法使萃取得到的有机相浓缩,向浓缩产物中加入100ml水,得到粗品;向粗品中加入10%氢氧化钠水溶液,再加入50mL甲基叔丁基醚洗涤两次后过滤,利用盐酸使溶液的pH为1.0,通过抽滤、烘干得到15.79g第三产物,其结构为2,4,6-三氟苯甲酸,HPLC纯度为99.3%,摩尔收率为96.2%,三步反应总摩尔收率为89.7%。
[1]柴文玉. 一种2,4,6-三氟苯甲酸的制备方法[P]. 黑龙江省:CN202310767966.7,2023-11-10.
三苯基硼烷,英文名:triphenylborane,CAS号:960-71-4,分子量:242.12300,密度:0.898 g/mL at 25°C,沸点:65-67°C,分子式:C18H15B,熔点:145°C(lit.),闪点:1°F,白色晶体,常温常压下稳定,避免氧化物、水分、空气接触,储存在干爽的惰性气体下,保持容器密封,储存在阴凉,干燥的地方。

方法一:以溴苯为原料,-78℃条件下,用n-BuLi拔溴得到苯基锂,再滴加三氯化硼的正己烷溶液,最后得到终产物三苯基硼烷。但是该路线费用较昂贵,正丁基锂、三氯化硼价格都较贵,由于在低温下反应(-78℃),反应条件苛刻,成本高,产率较低。正己烷、乙醚的使用也增加了反应的危险性。
方法二:以溴苯为原料,乙醚作为溶剂,在Mg的作用下,得到苯基溴化镁,再滴加三氟化硼的乙醚溶液,最后得到终产物三苯基硼烷。但是,乙醚闪点极低(-45℃)、挥发性强,容易发生爆炸,也降低了反应的安全性;低温反应(-78℃)也增加了成本;此反应操作也较繁琐。
方法三:以苯为原料,在室温条件下,用乙基溴化镁拔氢得到苯基溴化镁,再滴加三氟化硼的甲苯溶液,最后得到终产物三苯基硼烷。该路线异丙基溴化镁价格要比异丙基氯化镁贵67%。此路线所用的溶剂四氢呋喃对生殖系统具有毒性。
1、专利CN97193225.5公开了一种对于腔肠动物如薮枝螅和水螅、贝类如蔓足动物、紫贻贝和牡蛎,管栖多毛环节动物如盘管虫、龙介虫、无襟毛虫和被绕右旋虫或其它水生生物体的附着具有优异防污染淤塞作用的三苯基硼烷-松香胺加成物。该类化合物通过三苯基硼烷的氢氧化钠加成物和松香胺在水溶液中室温反应得到[2].
2、专利CN202111376565.6提供了一种光热剂,在氮气氛围下,将1?乙酰吲哚?3?酮、反应物Ar?CHO和反应介质混匀后加入碱,在20~40℃条件下反应10~20h,加入蒸馏水抽滤,干燥,加入反应甲苯,混匀后加入三苯基硼烷,加热回流,配位反应4~6小时,得到具有式(B)所示结构的光热剂。其母核结构含有吲哚?3?酮单元,利用商业化的原料吲哚?3?酮与芳基醛一锅煮反应,高产率获得一类具有刚性结构,吸收光谱红移,荧光量子产率降低,光热效率高的新型母核衍生物,可用于光热治疗癌症[3].
[1]浙江卫星石化股份有限公司. 一种无水三(五氟苯基)硼烷的制备方法:CN202010672806.0[P]. 2020-10-13.
[2]吉富精细化学品株式会社. 三苯基硼烷-松香胺加成物及其应用:CN97193225.5[P]. 1999-04-14.
[3]杭州师范大学,四川青木制药有限公司. 一种光热剂及其制备方法和应用:CN202111376565.6[P]. 2022-01-28.
探讨如何合成6-氟烟酸不仅有助于深入了解其合成途径和反应机理,还可以为其在药物合成等领域的应用提供重要参考。
简述:6-氟烟酸,英文名:6-Fluoronicotinic acid,CAS编号403-45-2,为白色固体,是合成含氟吡啶环抗菌药物的重要中间体。目前关于该化合物的制备方法文献报道较少,主要是以2-氟-5-甲基吡啶为原料,经高温氧化反应制备6-氟烟酸(参考文献:Journal of Med Chem,1990,33,1667-1675、US2002/10185、US5583148)。该方法需添加大量水用于溶解高锰酸钾,反应收率仅约为45%,同时会产生大量二氧化锰等重金属废物,存在废物处理困难、产能低、安全隐患等问题,不利于工业化规模生产。
合成优化:
利用2,5-二溴吡啶作为起始原料,在催化剂存在下,通过异丙基氯化镁格氏试剂的催化交换,与氯甲酸酯反应高度选择性地合成6-溴烟酸酯;随后,6-溴烟酸酯与四甲基氟化铵进行氟化反应,再经水解制备得到6-氟烟酸。这种制备方法环境友好、成本低、操作条件易于控制,适合工业化生产。具体实验步骤如下:
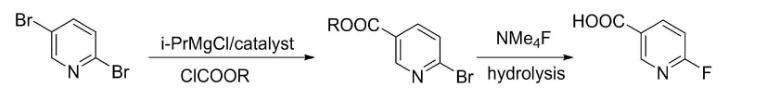
(1)氮气保护下,向反应瓶中加入400mL四氢呋喃和2,5-二溴吡啶(59.2g,0.25mol),控温-10~0℃下滴加异丙基氯化镁的四氢呋喃溶液(2.0M,150mL,0.30mol),滴毕保温反应3小时,HPLC中控原料2,5-二溴吡啶<0.5%,保温下继续加入碘化亚铜(1.43g,7.5mmol),然后滴加氯甲酸甲酯(28.4g,0.30mol),滴毕,室温搅拌1小时,HPLC中控中间体反应完全,6-溴烟酸甲酯88.6%、异构体5-溴吡啶-2-羧酸甲酯7.2%,0~10℃下滴加3MHCl调体系pH=3-4,加入乙酸乙酯萃取(200mL×3),有机层合并,饱和食盐水洗,有机层减压浓缩,加入100mL正庚烷/甲基叔丁基醚(体积比6/1)打浆,过滤得类白色固体6-溴烟酸甲酯42.9g,HPLC:97.1%,收率79.3%。
(2)向反应瓶中加入200mL甲苯和四甲基氟化铵(37.2g,0.40mol),加热至回流,利用分水器分出少量水至反应瓶内液体含水<300ppm,降温,减压浓缩出甲苯。加入6-溴烟酸甲酯(42.9g,0.20mol)和300mL DMF,升温至40-50℃反应16小时,HPLC中控原料反应完全,降至室温,减压蒸出DMF,20-30℃下滴加60g 30%NaOH溶液,滴毕,室温搅拌3小时,HPLC中控水解完全,6-氟烟酸钠94.6%、6-羟基烟酸钠5.0%,滴加20%盐酸调pH=3-4,过滤,滤饼用60mL水淋洗,真空干燥得白色固体6-氟烟酸24.1g,HPLC:98.0%,收率85.6%,mp:146.1-148.2℃。
参考文献:
[1]大连双硼医药化工有限公司. 一种6-氟烟酸的制备方法. 2022-12-06.
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手























