深入探究盐酸阿那格雷的合成方法对于提高生产效率和确保产品质量具有重要意义。
简介:盐酸阿那格雷,英文名称“Anagrelidehydrochloride”,商品名“安归宁”,分子式:C10H7Cl2N3O·HCl·H2O,分子量:310.56,化学名:6,7-二氯-1,5-二氢咪唑并[2,1-b]喹唑啉-2(3H)-酮单盐酸盐一水合物,6,7-dichloro-1,5-dihydroimidazo[2,1-b]-quinazolin-2(3H)-onemonohydrochloridemonohydrate。阿那格雷又称氯喹咪唑酮,为咪唑喹哪唑琳的衍生物,可以使红细胞的生成受到抑制,是一种降血小板药,对硫酸羟脲等产生抗药性的病人可采用阿那格雷治疗。一般使用阿那格雷盐酸盐,口服容易吸收,在体内多处代谢后随尿液流出。有降低血小板的生成和抵制血小板的聚集两种重要作用,以此降低血栓形成的隐患,适用于骨髓增生性疾病,如原发性血小板增多症与真性红细胞增多症,长期服用有轻微短暂的贫血现象,也会发生充血性心力衰竭和外源性过敏性肺泡炎等。
合成:
用间氯苯胺、水合氯醛和盐酸羟胺经缩合、环合、氯代及氧化反应制得6-氨基-2,3-二氯苯甲酸,再经环合、氯代、还原、取代、环合及成盐等反应制得抗凝血药盐酸阿那格雷,总收率约6.6%(以间氯苯胺计)。具体实验步骤如下:
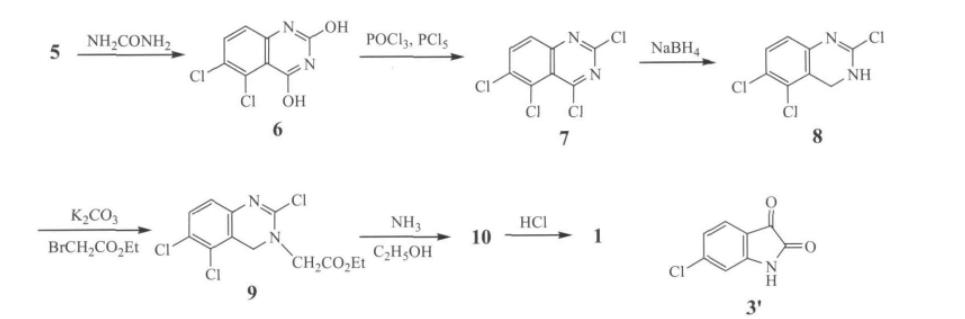
(1)N-(肟基乙酰)间氯苯胺(2)
将水合氯醛(100.0克,0.6摩尔)、间氯苯胺(56毫升,0.54摩尔)、盐酸羟胺(120克,1.73摩尔)、水(2.12升)、浓盐酸(50毫升)和无水硫酸钠(622克,4.4摩尔)按顺序加入反应瓶中,加热至沸腾,搅拌反应30分钟。冷却至室温后,用乙酸乙酯(500毫升×3)进行萃取,将有机相合并,经过无水硫酸镁干燥后过滤,滤液经减压浓缩,得到黄色固体2(100克,收率93.3%),熔点为177~179摄氏度。
(2)4-氯靛红(3)
将浓硫酸(750ml)加至反应瓶中,加热至80~85℃,搅拌下60min内分批加入2(150g,0.756mol)。同温反应15min,混合物倒入碎冰(2kg)中,生成红色沉淀。过滤,滤饼中加入水(200ml),搅拌下加入3mol/L氢氧化钠溶液(400ml),过滤,滤液于剧烈搅拌下加入浓盐酸(约50ml),当开始出现沉淀时(约pH8)再加入浓盐酸(30ml)和水(150ml),生成沉淀(约pH5)。过滤,滤饼干燥,所得红色粉末状3粗品(70g)用20%冰乙酸溶液重结晶,得红色针状晶体3(56g,43.75%),mp256~258℃,纯度98.8%。
(3)4,5-二氯靛红(4)
将3(75g,0.41mol)加至冰乙酸(1.5L)、磺酰氯(70ml,2.1mol)和少许碘的混合物中,于50℃搅拌4h,冷却至室温,过滤,得红色针状结晶4(49g,55.3%),mp244~245℃。
(4)6-氨基-2,3-二氯苯甲酸(5)
将4(75g,0.35mol)溶于5%氢氧化钠溶液(750ml)中,于20min内滴加30%双氧水(111ml,0.35mol),滴完搅拌30min。过滤,滤液中加入浓盐酸(约90ml)调至约pH4,产生沉淀。过滤,滤饼干燥后得黄色粉末状固体5(61g,85%),mp165~166℃。
(5)2,4-二羟基-5,6-二氯喹唑啉(6)
将5(70g,0.324mol)和尿素(210g,3.5mol)加至冰乙酸(1L)中,于120℃回流反应2h,冷却后过滤,得灰黄色固体6(63g,84.0%),mp338~340℃。
(6)2,4,5,6-四氯喹唑啉(7)
将6(45克,0.19摩尔)、五氯化磷(150克,3.8摩尔)和三氯氧磷(100毫升,5.7摩尔)加热至120摄氏度回流反应4小时。随后减压浓缩至体系黏稠,加入碎冰(500克),搅拌30分钟后进行过滤。将滤饼加入乙酸乙酯(2升)中,依次用5%碳酸钠溶液(2升)和饱和氯化钠溶液(2升)进行洗涤,经无水硫酸镁干燥后过滤,滤液减压浓缩,得到淡黄色固体7(40克,收率78.6%),熔点为309~310摄氏度。
(7)2,5,6-三氯-3,4-二氢喹唑啉(8)
将7(35g,0.13mol)溶于氯仿(350ml)和乙醇(140ml)的混合液中,冰浴条件下分批加入硼氢化钠(35g,0.93mol),常温反应2h,蒸除溶剂,残留物用50%乙醇(200ml)打浆洗涤,过滤,干燥后得白色固体8(30g,98.6%),mp314~316℃。
(8)2,5,6-三氯-3,4-二氢喹唑啉-3-乙酸乙酯(9)
将8(25g,0.11mol)、溴乙酸乙酯(20g,0.12mol)和碳酸钾(45g,0.33mol)加至丙酮(500ml)中,加热回流反应3h,冷却后过滤,滤液减压浓缩,残留物中加入乙醚-石油醚(3∶1,100ml)混合液,过滤,干燥后得淡黄色固体9(20.3g,57.7%),mp112~113℃。
(9)盐酸阿那格雷(1)
将9(20克,0.0625摩尔)和10%氨气的乙醇溶液(200毫升)注入密闭的耐压反应釜中,在120摄氏度条件下反应16小时。待反应结束后,冷却至室温,进行过滤。将得到的白色固体10加入到乙醇(560毫升)中,加热至回流,加入浓盐酸(16毫升),再次进行过滤。将滤液加热至沸腾,使用活性炭(1克)进行脱色处理,然后进行过滤。待滤液冷却至室温后,加入正庚烷(380毫升),室温条件下搅拌2小时后再次过滤,将滤饼干燥,得到白色固体1(14.6克,收率91.3%),熔点为280~282摄氏度。
参考文献:
[1]马鸿伟. 盐酸阿那格雷胶囊人体药代动力学研究[D]. 吉林大学, 2014.
[2]翁志洁,李建其,周斌. 盐酸阿那格雷的合成 [J]. 中国医药工业杂志, 2012, 43 (06): 405-408.
2,3,5,6-四氟-7,7',8,8'-四氰二甲基对苯醌作为一种重要的化合物,在许多领域都有广泛的应用,本文将介绍其不同用途,通过深入探讨2,3,5,6-四氟-7,7',8,8'-四氰二甲基对苯醌的多种用途,以期为读者呈现其广泛的应用前景。
简述:2,3,5,6-四氟-7,7',8,8'-四氰二甲基对苯醌,英文名称:2-[4-(dicyanomethylidene)-2,3,5,6-tetrafluorocyclohexa-2,5-dien-1-ylidene]propanedinitrile,CAS:29261-33-4,分子式:C12F4N4,外观与性状:黄色至橙色至棕色粉末,晶体,结晶粉末和/或大块。
应用:
1. 修复VO2薄膜表面氧缺陷
VO2表面氧缺陷的存在对VO2材料具有显著的电子掺杂效应,极大地影响材料的本征电子结构和相变性质.通过2,3,5,6-四氟-7,7’,8,8’-四氰二甲基对苯醌(F4TCNQ)分子表面吸附反应,可以有效消除表面 氧缺陷及其电子掺杂效应.利用同步辐射光电子能谱和X射线吸收谱原位研究了修复过程中电子结构的变 化以及界面的化学反应,发现这种方式使得VO2薄膜样品氩刻后得到的V3 +失去电子成功地被氧化成原先的V4 +,同时F4TCNQ分子吸附引起电子由衬底向分子层转移,界面形成带负电荷的分子离子物种.受电化 学性质的制约,F4TCNQ分子吸附反应修复氧缺陷较氧气氛退火更安全有效,不会引起表面过度氧化形成 V2O5。
2. 制备出反向有机发光二极管器件
首先,MoO3掺杂4,4′-[N-(1-萘基)-N-苯基-氨基]联苯(NPB:Mo O3质量比为2:1)与MoO3掺杂4,4’-双(N-二咔唑)-2,2’联苯(CBP:MoO3质量比为2:1)作为p型掺杂的空穴传输层分别使用在反向有机发光二极管(IOLEDs)中。由于CBP:Mo O3 到NPB的空穴传输能垒小于NPB:MoO3到NPB的空穴传输能垒,并且NPB:MoO3的电导率大于 CBP:Mo O3,使得NPB/ 10 nm CBP:MoO3/ 10 nm NPB:MoO3的双p掺杂层结构相比于 NPB/ 20 nm NPB:MoO3和NPB/ 20 nm CBP:MoO3结构提高了器件性能。
其次,为了获得更好的掺杂效果,研究人员采用MoO3掺杂NPB(NPB:MoO3)和2,3,5,6- 四氟-7,7',8,8'-四氰二甲基对苯醌掺杂NPB(NPB:F4-TCNQ)组成的杂化p型掺杂空穴传输层制备出反向有机发光二极管器件。与20 nm NPB:MoO3/ Al结构相比10 nm NPB:F4-TCNQ/ 10 nm NPB:MoO3/ Al结构使器件性能得到了提高。
3. 改进基于FASnI3 钙钛矿的太阳能电池的光电转化效率
有研究以甲脒锡碘(FASnI3)钙钛矿为研究对象,主要通过阳离子取代、优化钙钛矿成膜工艺以及空穴传输层(HTL)能级修饰等方法,改进基于FASnI3 钙钛矿的太阳能电池的光电转化效率。
通过引入乙脒(AcA)有机阳离子修饰FASnI3钙钛矿材料,结合两次旋涂钙钛矿溶液工艺,可以有效提升钙钛矿的膜层质量。在优化了AcA阳离子的掺杂浓度后,制备了基于AcA0 .1FA 0.9SnI 3 钙钛矿的最佳太阳能电池,其光电转化效率达到7.04%。
最后,以AcA0 .1FA 0.9SnI 3 钙钛矿为研究对象,通过向聚(3,4-乙烯二氧噻吩):聚苯乙烯磺酸(PEDOT:PSS)空穴传输层中引入2,3,5,6-四氟-7,7',8,8'-四氰二甲基对苯醌(F4-TCNQ),达到调节PEDOT:PSS能级的目的。修饰后的PEDOT:PSS 与AcA0 .1FA 0.9SnI3 钙钛矿的最高已占据分子轨道(HOMO)能级更加匹配,降低了空穴载流子在空穴传输层界面的能量损失,从而有效提升器件内部的电荷传输。最终,基于AcA0 .1FA 0.9SnI3 钙钛矿太阳能电池的最佳光电转化效率达到了7.84%,开路电压(VO C )达到0.55 V。
4. 提高有机-无机杂化钙钛矿太阳能电池(PSCs)的效率
为提高PSCs的效率,有研究以氟化SFX为核,4,4'-二甲氧基二苯胺为外围取代基团,通过两步反应合成了化合物2p F-X59和2m F X59。对性能更优异的2m F-X59基PSCs器件进一步优化。通过加入化合物2,3,5,6-四氟-7,7',8,8'-四氰二甲基对苯醌(F4TCNQ)进一步降低2m F-X59的HOMO能级并改善其薄膜形貌后,基于4wt% F4TCNQ优化的2m F-X59电池器件效率提高到18.13%。稳定性测试结果表明,不含敏感掺杂剂的2m F-X59基PSCs器件在空气中暴露超过500 h的 情况下能保持95%的初始效率,显示出很好的长期稳定性。
参考文献:
[1]许新. 基于有机阳离子修饰的FASnI_3钙钛矿太阳能电池研究[D]. 南京邮电大学, 2020. DOI:10.27251/d.cnki.gnjdc.2020.000974
[2]吴敏. 引入S,F元素制备SFX基空穴传输材料用于非敏感掺杂钙钛矿太阳能电池的研究[D]. 太原理工大学, 2019.
[3]靳松. 杂化p型掺杂的有机发光二极管[D]. 河北工业大学, 2016.
[4]王凯,张文华,刘凌云等. VO_2薄膜表面氧缺陷的修复:F_4TCNQ分子吸附反应 [J]. 物理学报, 2016, 65 (08): 351-357.
引言:
四唑具有重要的化学结构特点和广泛的应用领域。四唑化合物在医药、材料科学等领域具有重要的意义,其独特的结构和性质使其成为许多研究和应用的焦点。四唑的合成方法多样,通过不同的合成途径,可以合成出具有不同结构和性质的四唑衍生物,为各个领域的研究和应用提供了更多的可能性。本文将介绍四唑的合成方法,探讨不同的合成方法和其在四唑化合物研究中的意义,希望通过深入了解四唑的合成过程,能够更好地认识和应用这一类化合物,推动相关领域的发展和创新。
1. 什么是四唑?
四唑是一类人工合成的有机杂环化合物,由4个氮原子和1个碳原子组成5元环。四唑这个名字也指化学式为CH2N4的母体化合物,其三种同分异构体可以组成。化学式为CH2N4的母体化合物的结构如下图所示:
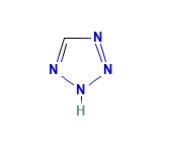
四唑是一类由1个碳原子和4个氮原子组成的双不饱和五元环芳香族杂环。它们在自然界中不存在。有趣的是,它们在稳定的杂环中具有最多的氮原子,因为五唑即使在低温下也是高度易爆的化合物1885年,瑞典化学家J. a. Bladin在乌普萨拉大学首次报道了四唑衍生物的合成。他观察到,二氰苯基肼和亚硝酸的反应形成了一种化合物,他后来为这种新的环结构命名为“四唑”。根据取代基的数量,四唑可以分为非取代基、单取代基、二取代基和三取代基。具有6π电子的5-取代四唑可能以I或II的互变异构体形式存在(如下图)。在溶液中,1H互变异构体是主要形式,但在气相中,2H互变异构体更稳定。

四唑衍生物是杂环的主要类别,对药用非常重要化学和药物设计不仅因为它们对羧酸的生物等分体酸和酰胺部分,以及它们的代谢稳定性和有益的理化性质。有超过20个 FDA 批准的药物含有1H-或 2H-四唑取代基。
2. 四唑是如何形成的?
四唑通常是通过有机腈和叠氮化钠(NaN3)在催化剂的作用下反应而形成的。下面是这个过程的一个分解:
(1)腈激活:催化剂,如碘或二氧化硅负载的亚硫酸氢钠,为反应准备腈分子。它使腈中的碳氮三键更容易被电子吸引,为下一步做准备。
(2)叠氮化物攻击:随着腈的激发,叠氮化物离子(N3),像亲核试剂(亲电子)一样,攻击腈中带正电荷的碳原子,形成一个新的键。
(3)环形成(环化):生成的中间体发生环化反应。想象一下叠氮化物基团的末端氮向后延伸并与连接到另外两个氮的碳原子成键。这就产生了具有四个氮和一个中心碳的标志性五元四唑环。
(4)质子化:最后一步涉及一个质子(H+)从水分子或酸连接到四唑环。这种质子化完成了四唑分子的形成,通常是一个5-取代的1H-四唑。
3. 了解四唑合成机理
合成四唑的最常见途径是叠氮化物与腈、异氰化物和亚胺基官能团等合成子之间的环加成反应。在这些方法中,将酰胺转化为四唑是必不可少的方法之一,因为酰胺是市售的,也可以毫无困难地制备。以前的大多数酰胺报告都有一些缺点,例如分离酰胺酰氯、叠氮化前去除酰氯以及使用昂贵的试剂,如四氯化硅和叠氮化硅烷。R Sribalan等人的报道中,专注于改进方法,无需分离中间体,并使用更便宜的试剂从酰胺原位合成四唑。在此基础上,提出了四氮唑形成的合成机理,如下图所示:
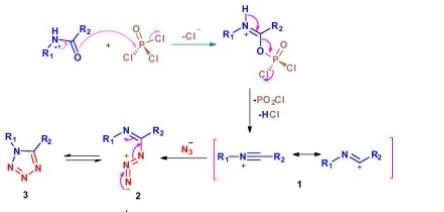
最初,在POCl3存在下,酰胺可以像Bischler Napieralski反应一样,在消除氯化氢的情况下转化为腈中间体。叠氮化物与氮离子的亲核性加成得到中间体2,随后分子内环化生成四唑3。根据提出的机理,我们了解到选择性取决于叠氮-四唑平衡。随着叠氮化钠的化学计量量的增加,平衡向四氮唑3的形成转移。同时,减少叠氮化钠的用量可能不利于四氮唑的生成。合成四唑的一般程序如下:
(1)热方法
在双颈圆底烧瓶中分别添加苯甲苯胺(1.01 mmol)、三氯氧磷(10.15 mmol)和叠氮化钠(4.06 mmol)。烧瓶的一个颈部与带保护管的回流冷凝器连接,另一个颈部与氮气入口连接。在80℃下在氮气氛下将反应混合物搅拌9 h。然后将它冷却,用冰水小心地淬灭,用饱和的碳酸氢钠溶液中和。用乙酸乙酯(75 mL)萃取产物,用水(2 × 75 mL)和盐水(75 mL)洗涤。分离有机层并且经无水Na2SO4干燥并且在低压下浓缩。
(2)微波照射法
在10 mL双颈RB烧瓶中添加苯甲苯胺(1.01 mmol)、三氯氧磷(10.15 mmol)和叠氮化钠(4.04 mmol)。然后将烧瓶的一个颈部与带保护管的回流冷凝器连接,另一个颈部与氮气入口连接。将反应混合物中的氮气吹扫10 min。然后移除氮气入口并且用塞子突然关闭。反应装置固定在微波合成器中(较好地避免了密封管的微波照射)。将反应混合物在120 W、80℃下进行6 min的微波辐照。然后使反应混合物达到室温。在冷却时,将反应混合物用碎冰淬火并用碳酸氢钠溶液中和。用乙酸乙酯(75 mL)萃取产物,用水(2 × 75 mL)和盐水(75 mL)洗涤。分离有机层并且经无水Na2SO4干燥并且在低压下浓缩。
4. 探索四唑合成的名称反应
合成四唑没有一种特定的反应,因为制造五元杂环的方法有很多种。然而,一些常见的方法包括:
(1)Curtius重排:该反应涉及酰基叠氮化物的热分解,其重排形成异氰酸酯,然后环化形成四唑。
(2)施密特反应:该反应涉及醛或酮与腙酸(HN3)处理,形成中间亚胺,然后环化形成四唑。
(3)Hantzsch反应:该反应涉及腈、叠氮钠(NaN3)和氯化铵(NH4Cl)在酸性条件下缩合形成四唑。
(4)使用铜催化点击化学:该方法使用有机叠氮化物和末端炔来形成具有高区域选择性和产率的1,5-二取代四唑。
5. 纯四唑合成的实用方法
腈(10 mmol),NaN3(12 mmol,0.78g),和Py·HCl(10mmol,1.15g)在20mL DMF中的溶液加入50mL圆底烧瓶中。反应混合物在110℃下加热8h,剧烈搅拌。采用高效液相色谱法和薄层色谱法监测转化情况。将反应混合物冷却至室温,溶解于4mLNa0H水溶液(5 M)中,搅拌30 min。通过去除DMF和Py,在减压下对溶液进行浓缩;将反应残渣溶解于10ml水中。用HCI(3M,10 mL)调节pH值为1,形成沉淀。然后过滤沉淀,用2x10m的3M盐酸洗涤,在80℃下干燥过夜,得到纯四氮唑,为白色固体(1.23g)。产率 84%(熔点 216-218℃)。
6. 结论:拥抱四唑的潜力
通过本文对四唑的合成进行探讨,我们不仅了解了四唑化合物的意义和重要性,还深入探讨了其在各个领域中的广泛应用。四唑的合成方法多样化,为研究人员提供了更多的选择和可能性。四唑化合物的独特结构和性质为各行业带来了新的机遇和挑战,促进了相关领域的发展和创新。鉴于四唑化合物的潜力和重要性,我们呼吁研究人员继续深入探索四唑的化学特性和应用领域,拥抱其潜力,推动科学技术的进步,为人类社会的发展做出更大的贡献。让我们共同努力,拓展四唑的应用领域,开创更加美好的未来。
参考:
[1]Sribalan R, Lavanya A, Kirubavathi M, et al. Selective synthesis of ureas and tetrazoles from amides controlled by experimental conditions using conventional and microwave irradiation[J]. Journal of Saudi Chemical Society, 2018, 22(2): 198-207.
[2]Zhou Y, Yao C, Ni R, et al. Amine Salt–Catalyzed Synthesis of 5-Substituted 1 H-Tetrazoles from Nitriles[J]. Synthetic Communications®, 2010, 40(17): 2624-2632.
[3]https://en.wikipedia.org/wiki/Tetrazole
[4]https://pubchem.ncbi.nlm.nih.gov/compound/67519
[5]https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6376451
2,4-二甲氧基苯甲醛是一种重要的化合物,其合成方法备受关注。本文将介绍2,4-二甲氧基苯甲醛的合成方法,以供相关研究人员参考。
背景:2,4-二甲氧基苯甲醛是重要的有机中间体之一,广泛用于医药、农药、染料等的合成,如用于对金黄色葡萄球菌、大肠杆菌等有很强抑制作用的coumerungin、当归内酯等香豆素类化合物的合成。
合成:
1. 方法一:
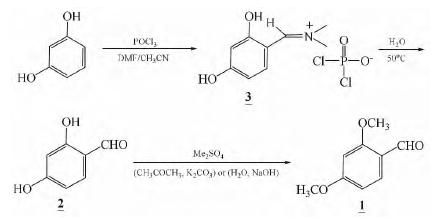
(1)2,4-二羟基苯甲醛的制备
将49.3 g(0.675 mol)N,N-二甲基甲酰胺 (DMF)和150 mL乙腈混合搅拌,后将88.16 g (0.575 mol)三氯氧磷滴入该溶液,在室温下反应 1 h,将溶液冷却至-14~-17℃加入55.06 g(0.5 mol)间苯二酚和150 mL乙腈,反应2 h,然后再在室温下反应1 h,抽滤得到微粉色的粉末,得到的粉末分3次加水共340 mL,在52℃反应0.5 h,放置室温加入1~2 mL硫代硫酸钠,在5℃下反应2 h,抽滤,用冷水洗涤,30℃真空干燥得24.3 g 2-4-二 羟基苯甲醛白色粉末,收率为70.4%,m.p.134.5~ 136.4℃。
(2)2,4-二甲氧基苯甲醛的制备
在50 mL的三颈瓶中,加入3.5 g(0.025 mol)2,4-二羟基苯甲醛,3.2 g(0.025 mol)硫酸二甲酯,1 g(0.05 mol)水,3 g(0.075 mol)氢氧化 钠,搅拌1~2 h,旋干,以v(乙酸乙酯)∶v(环己烷)=3∶7进行硅胶柱层析纯化得到0.189 g产物,收率为4.5%,m.p.67.2~69.3℃。
2. 方法二 2.3.2.1 1,3-二甲氧基苯的制备
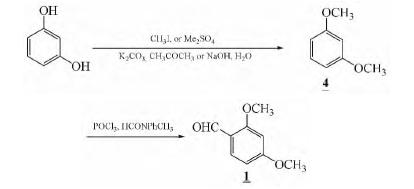
(1)硫酸二甲酯做甲基化试剂的合成方法
反应器中加入24 g(0.6 mol)氢氧化钠,7 mL(0.2 mol)水和22 g(0.2 mol)间苯二酚在100℃下溶解后,滴加25 g(0.2 mol)硫酸二甲酯,反应5 h,用水冲洗分出油层,水层用二氯甲烷提取,油层和萃取液合并,水洗,用无水硫酸镁干燥,用水浴加热回收二氯甲烷,得到22 g产物,收率为79.7%。
(2)甲基碘做甲基化试剂的合成方法
在配有回流冷凝管和氯化钙干燥管的圆底烧瓶中按1∶10(反应物质量与溶剂体积)的比例放入间苯二酚27.5 g (0.25 mol)与275 mL丙酮,在蒸汽浴加热溶解, 放入35 g(0.25 mol)无水碳酸钾,和86.5 g(0.61 mol)甲基碘,在水浴上回流在60~70℃下反应 6 h,用硫酸酸化,用冷水冷却至室温,将所得混 合物在蒸汽浴下蒸馏,除去丙酮,再进行减压蒸馏 无油状物馏出,通过洗滤分离,用冷水洗涤2次, 并干燥得到10.6 g淡黄色液体,收率为30.7%。
(3) 2,4-二甲氧基苯甲醛的制备
反应器中加入4.8 mL(0.053 mol)三氯氧磷和 6.5 mL(0.053 mol)N-甲基甲酰苯胺,放置45 min 后,25℃下滴加7 mL(0.053 mol)1,3-二甲氧基苯,在34℃下反应3 h,放置过夜,慢慢倒入135 mL 冷水中,抽滤收集固体,用15 mL二氯甲烷溶解,加2 g活性炭回流1 h,热滤去除活性碳,将滤液在蒸馏回收二氯甲烷得到5.6 g淡黄色粉末,收率 63.6%,m.p.66.2~67.5℃。
3. 方法三:
称取DMF21.9 g(0.30 mol)投入四口烧瓶中,然后在冰浴条件下缓慢滴加25.9 g三氯氧磷(0.17 mol),滴加45 min,在滴加时温度稳定,然后反应30 min;升温至30℃,在温度稳定的情况下缓慢滴加 20.7 g 1,3-二甲氧基苯(0.15 mol),滴加1 h后恒 温反应3 h,然后将反应液静置过夜,倒入600 m L冰水中,用NaOH溶液中和到pH为7,搅拌2 h,然后减压过滤,烘干,得白色针状固体,真空干燥,得到固体17.41 g。HPLC测定其纯度为98.60%,收率为 75.30%,mp为71.6~72.0℃。
参考文献:
[1]马成,热娜·卡斯木,阿孜古丽·买买提尼亚孜等. 基于间苯二酚合成2,4-二甲氧基苯甲醛 [J]. 精细化工中间体, 2014, 44 (01): 19-21. DOI:10.19342/j.cnki.issn.1009-9212.2014.01.005
[2]沈立,金宁人,张建庭等. 光学材料中间体DADMNS的合成研究 [J]. 现代化工, 2012, 32 (06): 64-67. DOI:10.16606/j.cnki.issn0253-4320.2012.06.024
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手























