你好,最后您解决了吗,是怎么处理了呢?
你好,最后您解决了吗,是怎么处理了呢?
易燃易爆介质垫片是否可以用非金属?那个规范要求?如果用非金属无法导出静电。
易燃易爆介质垫片是否可以用非金属?那个规范要求?如果用非金属无法导出静电。
都有什么化工设备呢,看看有没有熟悉的
显示全部都有什么化工设备呢,看看有没有熟悉的
 1
1
丙酸诺龙作为一种常见的蛋白同化激素,其残留对人体健康构成严重威胁。本文旨在深入探讨丙酸诺龙残留的检测技术及控制策略,以期为食品安全监管提供科学依据。
简述:
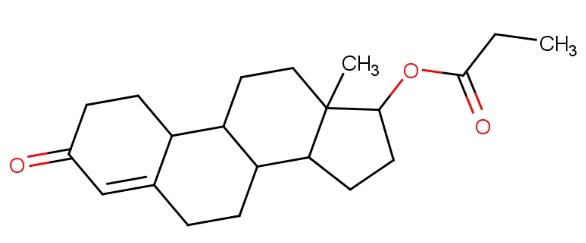
丙酸诺龙(英语:Nandrolone propionate,或称为19-去甲睾酮-17β-丙酸酯 19-nortestosterone 17β-propionate,商品名为Anabolicus)是一种雄激素同化类固醇(AAS)药物,在西班牙有出售。19-去甲睾酮(Nandrolone,诺龙),丙酸诺龙的最终活性形式。丙酸诺龙的结构如下:
背景:
蛋白同化激素,亦称为合成代谢类固醇(Anabolic-androgenic steroids, AAS),是一类外源性雄性激素,具有显著的蛋白同化作用,能够在一定时间内促使体内蛋白质的积累。在过去50年中,由于其能促进动物生长、提升饲料转化率以及对育种的积极影响,蛋白同化激素在肉牛养殖业中得到了广泛应用。然而,这些激素的使用可能导致男性精子生成减少、睾丸萎缩、不孕以及不可逆的女性化乳房发育;女性则可能出现月经不规律、脱发或体毛增加、阴蒂肥大、乳房萎缩和声音变粗等问题。长期使用类固醇激素还可能对心血管系统和肝脏造成影响,增加高血压、动脉粥样硬化和肝功能损害的风险。此外,有研究指出,蛋白同化激素在体内的残留还可能引发细胞病变,增加癌症发生的风险。因此,为了保护消费者的健康,需要建立一种灵敏度高、特异性强且操作简便的检测方法,以有效监控蛋白同化激素的残留情况。
目前,常用的检测蛋白同化激素的方法有高效液相色谱法(HPLC)、超高液相色谱-质谱联用法(UPLC-MS)、气相色谱-质谱联用法(GC-MS)和免疫检测法等。
检测与控制蛋白同化激素的残留:
(1)报道一
张燕等人报道了一种检测蛋白同化激素(诺龙,群勃龙,丙酸诺龙,雄诺龙,美雄诺龙)的量子点标记免疫层析试纸条,包括样品垫a、硝酸纤维素膜b、吸水垫c和PVC背板,其特征在于,在PVC背板上按顺序依次粘附有样品垫a、硝酸纤维素膜b、吸水垫c;硝酸纤维素膜c上分别包被有诺龙抗原构成的检测线d和羊抗兔二抗构成的质控线e。
(2)报道二
张燕等人报道了一种快速检测食品中蛋白同化激素(诺龙,群勃龙,丙酸诺龙,雄诺龙,美雄诺龙)的免疫亲和凝胶检测柱的制备方法,以琼脂糖凝胶为固相载体,将诺龙抗体和溴化氰活化的琼脂糖凝胶偶联制备抗体胶作为检测层,将HRP抗体与溴化氰活化的琼脂糖凝胶偶联制备HRP抗体胶作为质控层,装入1mL的固相萃取柱,制备免疫亲和凝胶检测柱。该方法研制了一种新型快速定性半定量检测食品中诺龙、群勃龙、丙酸诺龙、雄诺龙和美雄诺龙的免疫亲和凝胶柱检测产品,检测限分别为5μg/L、20μg/L、20μg/L、40μg/L和60μg/L。
方法包括如下步骤:(1)封闭胶的制备;(2)被检物抗体胶的制备:(3)HRP抗体胶的制备;(4)质控层制备;由步骤(3)制得的HRP抗体胶与步骤(1)制得的封闭胶按照比例混合好后,取150μL混合胶加入到1mL的SPE塑料柱中,用注射器活塞将PBS压出;(5)检测层制备检测层是由抗体胶与步骤(1)制得的封闭胶按照比例混合好后,取150μL加入到1mL的SPE塑料柱中,用注射器活塞将PBS压出;(6)检测柱组装。
(3)报道三
王敬等人报道了一种风味牛肉饼中蛋白同化剂质控样品的制备方法,包括以下步骤:(1)分别制备丙酸诺龙、丙酸睾酮、美雄酮和睾酮标准溶液,将各个标准溶液稀释混合配制标准混合工作溶液;(2)风味牛肉饼样品绞碎1~3次得肉糜状样品,肉糜状样品与标准混合工作溶液混合,加水得样品匀浆;(3)样品匀浆预冷冻,然后真空冷冻干燥得冻干样品;(4)冻干样品粉碎。该制备方法保证了蛋白同化剂质控样品的均匀性和稳定性,且制备流程具有可推广性,对调理肉制品基质兽药残留的质控样制备具有指导意义。
参考:
[1] 石家庄海关技术中心. 一种风味牛肉饼中蛋白同化剂质控样品的制备方法. 2022-11-29.
[2] 天津科技大学. 一种检测蛋白同化激素的量子点标记免疫层析试纸条及其制备方法. 2017-02-22.
[3] 天津科技大学. 一种检测食品中蛋白同化激素的免疫亲和凝胶检测柱的制备方法. 2017-09-22.
[4]https://zh.wikipedia.org/wiki/%E4%B8%99%E9%85%B8%E8%AF%BA%E9%BE%99
丙酸诺龙作为一种常见的蛋白同化激素,其残留对人体健康构成严重威胁。本文旨在深入探讨丙酸诺龙残留的检测技术及控制策略,以期为食品安全监管提供科学依据。
简述:
丙酸诺龙(英语:Nandrolone propionate,或称为19-去甲睾酮-17β-丙酸酯 19-nortestosterone 17β-propionate,商品名为Anabolicus)是一种雄激素同化类固醇(AAS)药物,在西班牙有出售。19-去甲睾酮(Nandrolone,诺龙),丙酸诺龙的最终活性形式。丙酸诺龙的结构如下:
背景:
蛋白同化激素,亦称为合成代谢类固醇(Anabolic-androgenic steroids, AAS),是一类外源性雄性激素,具有显著的蛋白同化作用,能够在一定时间内促使体内蛋白质的积累。在过去50年中,由于其能促进动物生长、提升饲料转化率以及对育种的积极影响,蛋白同化激素在肉牛养殖业中得到了广泛应用。然而,这些激素的使用可能导致男性精子生成减少、睾丸萎缩、不孕以及不可逆的女性化乳房发育;女性则可能出现月经不规律、脱发或体毛增加、阴蒂肥大、乳房萎缩和声音变粗等问题。长期使用类固醇激素还可能对心血管系统和肝脏造成影响,增加高血压、动脉粥样硬化和肝功能损害的风险。此外,有研究指出,蛋白同化激素在体内的残留还可能引发细胞病变,增加癌症发生的风险。因此,为了保护消费者的健康,需要建立一种灵敏度高、特异性强且操作简便的检测方法,以有效监控蛋白同化激素的残留情况。
目前,常用的检测蛋白同化激素的方法有高效液相色谱法(HPLC)、超高液相色谱-质谱联用法(UPLC-MS)、气相色谱-质谱联用法(GC-MS)和免疫检测法等。
检测与控制蛋白同化激素的残留:
(1)报道一
张燕等人报道了一种检测蛋白同化激素(诺龙,群勃龙,丙酸诺龙,雄诺龙,美雄诺龙)的量子点标记免疫层析试纸条,包括样品垫a、硝酸纤维素膜b、吸水垫c和PVC背板,其特征在于,在PVC背板上按顺序依次粘附有样品垫a、硝酸纤维素膜b、吸水垫c;硝酸纤维素膜c上分别包被有诺龙抗原构成的检测线d和羊抗兔二抗构成的质控线e。
(2)报道二
张燕等人报道了一种快速检测食品中蛋白同化激素(诺龙,群勃龙,丙酸诺龙,雄诺龙,美雄诺龙)的免疫亲和凝胶检测柱的制备方法,以琼脂糖凝胶为固相载体,将诺龙抗体和溴化氰活化的琼脂糖凝胶偶联制备抗体胶作为检测层,将HRP抗体与溴化氰活化的琼脂糖凝胶偶联制备HRP抗体胶作为质控层,装入1mL的固相萃取柱,制备免疫亲和凝胶检测柱。该方法研制了一种新型快速定性半定量检测食品中诺龙、群勃龙、丙酸诺龙、雄诺龙和美雄诺龙的免疫亲和凝胶柱检测产品,检测限分别为5μg/L、20μg/L、20μg/L、40μg/L和60μg/L。
方法包括如下步骤:(1)封闭胶的制备;(2)被检物抗体胶的制备:(3)HRP抗体胶的制备;(4)质控层制备;由步骤(3)制得的HRP抗体胶与步骤(1)制得的封闭胶按照比例混合好后,取150μL混合胶加入到1mL的SPE塑料柱中,用注射器活塞将PBS压出;(5)检测层制备检测层是由抗体胶与步骤(1)制得的封闭胶按照比例混合好后,取150μL加入到1mL的SPE塑料柱中,用注射器活塞将PBS压出;(6)检测柱组装。
(3)报道三
王敬等人报道了一种风味牛肉饼中蛋白同化剂质控样品的制备方法,包括以下步骤:(1)分别制备丙酸诺龙、丙酸睾酮、美雄酮和睾酮标准溶液,将各个标准溶液稀释混合配制标准混合工作溶液;(2)风味牛肉饼样品绞碎1~3次得肉糜状样品,肉糜状样品与标准混合工作溶液混合,加水得样品匀浆;(3)样品匀浆预冷冻,然后真空冷冻干燥得冻干样品;(4)冻干样品粉碎。该制备方法保证了蛋白同化剂质控样品的均匀性和稳定性,且制备流程具有可推广性,对调理肉制品基质兽药残留的质控样制备具有指导意义。
参考:
[1] 石家庄海关技术中心. 一种风味牛肉饼中蛋白同化剂质控样品的制备方法. 2022-11-29.
[2] 天津科技大学. 一种检测蛋白同化激素的量子点标记免疫层析试纸条及其制备方法. 2017-02-22.
[3] 天津科技大学. 一种检测食品中蛋白同化激素的免疫亲和凝胶检测柱的制备方法. 2017-09-22.
[4]https://zh.wikipedia.org/wiki/%E4%B8%99%E9%85%B8%E8%AF%BA%E9%BE%99
我司提供欧洲进口 金属C型密封,O型密封、内部可填充弹簧,O型可以充入惰性气体,外表面涂层金、银、镍、PTFE等,适用于超低温、超高温、超高压等,温度-270℃至+1180℃,压力至680Mpa,主要应用于 压力容器、燃气轮机、试验仪器、石油石化等,需要联系 18678860293
显示全部我司提供欧洲进口 金属C型密封,O型密封、内部可填充弹簧,O型可以充入惰性气体,外表面涂层金、银、镍、PTFE等,适用于超低温、超高温、超高压等,温度-270℃至+1180℃,压力至680Mpa,主要应用于 压力容器、燃气轮机、试验仪器、石油石化等,需要联系 18678860293
1
引言:
N-(3-氯丙基)二丁基胺作为一种重要的有机化合物,其纯度对后续反应和产品性能有着至关重要的影响。因此,建立一种准确、灵敏、高效的杂质检测方法,对于保障该化合物的质量显得尤为重要。
简介:
N-(3-氯丙基)二丁基胺(N-(3-Chloropropyl)dibutylamine),其分子式为C11H24ClN,是一种无色至淡黄色的液体,能够很好地溶解于多种有机溶剂,并广泛用于医药中间体及有机溶剂。在其合成过程中,常会伴随生成二正丁胺、3-溴-1-丙醇、3-二丁氨基-1-丙醇和溴氯丙烷等副产物。作为盐酸决奈达隆的起始原料,N-(3-氯丙基)二丁基胺的纯度对于最终产品的质量至关重要,因此必须严格控制其杂质含量。N-(3-氯丙基)二丁基胺的结构如下:

杂质检测:
陈碧楚等人提供一种N-(3-氯丙基)二丁基胺中杂质的检测方法,包括以下步骤:
1. 溶液配制。
(1)空白溶液:甲醇。
(2)对照品贮备溶液:分别取适量的二正丁胺对照品、3-溴-1-丙醇对照品、3-二丁氨基-1-丙醇对照品和溴氯丙烷对照品,精密称定,用甲醇定量稀释溶解,制成每1mL中各含1000μg的混合溶液,得对照品贮备溶液。即对照品贮备溶液中,二正丁胺、3-溴-1-丙醇、3-二丁氨基-1-丙醇和溴氯丙烷的浓度均为1000μg/mL。
(3)对照品溶液:精密移取适量对照品贮备溶液,用甲醇定量稀释,制成每1mL中各含100μg的混合溶液,得对照品溶液。即对照品溶液中,二正丁胺、3-溴-1-丙醇、3-二丁氨基-1-丙醇和溴氯丙烷的浓度均为100μg/mL。
(4)供试品溶液:取200mg N-(3-氯丙基)二丁基胺样品,精密称定,置于10mL容量瓶中,加入甲醇溶解并稀释至刻度,得供试品溶液。
(5)加标供试品溶液:取200mg N-(3-氯丙基)二丁基胺样品,精密称定,置于10mL容量瓶中,精密加入上述对照品贮备溶液1mL,用甲醇溶解并定容至刻度,得加标供试品溶液。
2. 采用气相色谱法检测上述空白溶液、对照品溶液、供试品溶液和加标供试品溶液。具体气相色谱法的色谱条件包括:
(1)采用DB-624色谱柱(60m×0.53mm,5μm);
(2)气相色谱的升温程序为:起始温度50℃,维持5min,以20℃/min的速率升温至230℃,维持15min;
(3)检测器为氢火焰离子化(FID)检测器,进样口温度为250℃,检测器温度为280℃;
(4)载气为氮气,流速5mL/min;
(5)进样体积为1μL;
(6)分流比为10:1。
该N-(3-氯丙基)二丁基胺中杂质的检测方法,空白溶剂不干扰各成分的检测,各成分间的分离度符合要求,方法专属性良好。
参考:
[1]石家庄四药有限公司. 一种N-(3-氯丙基)二丁基胺中杂质的检测方法. 2024-06-21.
引言:
N-(3-氯丙基)二丁基胺作为一种重要的有机化合物,其纯度对后续反应和产品性能有着至关重要的影响。因此,建立一种准确、灵敏、高效的杂质检测方法,对于保障该化合物的质量显得尤为重要。
简介:
N-(3-氯丙基)二丁基胺(N-(3-Chloropropyl)dibutylamine),其分子式为C11H24ClN,是一种无色至淡黄色的液体,能够很好地溶解于多种有机溶剂,并广泛用于医药中间体及有机溶剂。在其合成过程中,常会伴随生成二正丁胺、3-溴-1-丙醇、3-二丁氨基-1-丙醇和溴氯丙烷等副产物。作为盐酸决奈达隆的起始原料,N-(3-氯丙基)二丁基胺的纯度对于最终产品的质量至关重要,因此必须严格控制其杂质含量。N-(3-氯丙基)二丁基胺的结构如下:
杂质检测:
陈碧楚等人提供一种N-(3-氯丙基)二丁基胺中杂质的检测方法,包括以下步骤:
1. 溶液配制。
(1)空白溶液:甲醇。
(2)对照品贮备溶液:分别取适量的二正丁胺对照品、3-溴-1-丙醇对照品、3-二丁氨基-1-丙醇对照品和溴氯丙烷对照品,精密称定,用甲醇定量稀释溶解,制成每1mL中各含1000μg的混合溶液,得对照品贮备溶液。即对照品贮备溶液中,二正丁胺、3-溴-1-丙醇、3-二丁氨基-1-丙醇和溴氯丙烷的浓度均为1000μg/mL。
(3)对照品溶液:精密移取适量对照品贮备溶液,用甲醇定量稀释,制成每1mL中各含100μg的混合溶液,得对照品溶液。即对照品溶液中,二正丁胺、3-溴-1-丙醇、3-二丁氨基-1-丙醇和溴氯丙烷的浓度均为100μg/mL。
(4)供试品溶液:取200mg N-(3-氯丙基)二丁基胺样品,精密称定,置于10mL容量瓶中,加入甲醇溶解并稀释至刻度,得供试品溶液。
(5)加标供试品溶液:取200mg N-(3-氯丙基)二丁基胺样品,精密称定,置于10mL容量瓶中,精密加入上述对照品贮备溶液1mL,用甲醇溶解并定容至刻度,得加标供试品溶液。
2. 采用气相色谱法检测上述空白溶液、对照品溶液、供试品溶液和加标供试品溶液。具体气相色谱法的色谱条件包括:
(1)采用DB-624色谱柱(60m×0.53mm,5μm);
(2)气相色谱的升温程序为:起始温度50℃,维持5min,以20℃/min的速率升温至230℃,维持15min;
(3)检测器为氢火焰离子化(FID)检测器,进样口温度为250℃,检测器温度为280℃;
(4)载气为氮气,流速5mL/min;
(5)进样体积为1μL;
(6)分流比为10:1。
该N-(3-氯丙基)二丁基胺中杂质的检测方法,空白溶剂不干扰各成分的检测,各成分间的分离度符合要求,方法专属性良好。
参考:
[1]石家庄四药有限公司. 一种N-(3-氯丙基)二丁基胺中杂质的检测方法. 2024-06-21.
1
引言:
精准定量是药物合成与质量控制的关键。 本研究旨在开发一种高效、灵敏的方法,用于检测4-苄氧基苯亚甲基-4-氟苯胺中微量杂质4-苄氧基苯亚甲基苯胺,以确保产品的纯度和安全性。
简介:
4-苄氧基苯亚甲基-4-氟苯胺作为合成依替米贝和依折麦布等药物的关键原料,是重要的医药中间体。然而,在合成过程中可能会生成杂质4-苄氧基苯亚甲基苯胺(SCR-5014),该杂质可能会在最终药物中富集,因此需要严格控制其含量。4-苄氧基苯亚甲基-4-氟苯胺和4-苄氧基苯亚甲基苯胺的结构式分别如下式I和式II所示:

杂质检测:
王霞等人报道了一种检测4-苄氧基苯亚甲基-4-氟苯胺中杂质4-苄氧基苯亚甲基苯胺的HPLC方法。在该方法中采用苯基键合硅胶色谱柱;流动相由含有胺类物质的水溶液或者含有胺类物质的磷酸盐、醋酸盐或甲酸盐缓冲液和有机溶剂组成;检测波长为250~350nm。使用该HPLC分析方法后,杂质4-苄氧基苯亚甲基苯胺检测灵敏度明显提高,与主成份及其他杂质分离度很好,可有效地控制产品质量。具体如下:
(1)色谱条件
仪器:Agilent1260HPLC
色谱柱:五氟苯基柱(4.6×100mm,2.6μm)
流动相A:磷酸盐缓冲液(称取磷酸二氢钾1.36g,加水1000ml使溶解,加入三乙胺5ml,磷酸调节pH6.8)
流动相B:乙腈:甲醇—1:1
流动相:流动相A-流动相B(40:60)
柱温:35℃
流速:1.0ml/min
检测波长:320nm
进样量:5ul
分析时间:记录色谱图至主成分峰保留时间的2倍
溶剂:乙腈
(2)样品配制
供试品溶液:取4-苄氧基苯亚甲基-4-氟苯胺适量,加溶剂制成每ml含4-苄氧基苯亚甲基-4-氟苯胺3.0mg的溶液,作为供试品溶液。
对照品溶液:取SCR-5014对照品适量,加溶剂制成每ml含SCR-5014约0.6μg/ml的溶液,作为对照品溶液。
(3)测定结果
检测结果表明SCR-5014与主峰及其他杂质分离度很好。
参考:
[1] 先声药业有限公司,江苏先声药业有限公司. 一种检测4-苄氧基苯亚甲基-4-氟苯胺中的4-苄氧基苯亚甲基苯胺的HPLC方法. 2021-06-29.
[2] 南京先声东元制药有限公司,江苏先声药业有限公司. 一种检测4-苄氧基苯亚甲基-4-氟苯胺中的4-苄氧基苯亚甲基苯胺的HPLC方法. 2017-11-17.
显示全部引言:
精准定量是药物合成与质量控制的关键。 本研究旨在开发一种高效、灵敏的方法,用于检测4-苄氧基苯亚甲基-4-氟苯胺中微量杂质4-苄氧基苯亚甲基苯胺,以确保产品的纯度和安全性。
简介:
4-苄氧基苯亚甲基-4-氟苯胺作为合成依替米贝和依折麦布等药物的关键原料,是重要的医药中间体。然而,在合成过程中可能会生成杂质4-苄氧基苯亚甲基苯胺(SCR-5014),该杂质可能会在最终药物中富集,因此需要严格控制其含量。4-苄氧基苯亚甲基-4-氟苯胺和4-苄氧基苯亚甲基苯胺的结构式分别如下式I和式II所示:
杂质检测:
王霞等人报道了一种检测4-苄氧基苯亚甲基-4-氟苯胺中杂质4-苄氧基苯亚甲基苯胺的HPLC方法。在该方法中采用苯基键合硅胶色谱柱;流动相由含有胺类物质的水溶液或者含有胺类物质的磷酸盐、醋酸盐或甲酸盐缓冲液和有机溶剂组成;检测波长为250~350nm。使用该HPLC分析方法后,杂质4-苄氧基苯亚甲基苯胺检测灵敏度明显提高,与主成份及其他杂质分离度很好,可有效地控制产品质量。具体如下:
(1)色谱条件
仪器:Agilent1260HPLC
色谱柱:五氟苯基柱(4.6×100mm,2.6μm)
流动相A:磷酸盐缓冲液(称取磷酸二氢钾1.36g,加水1000ml使溶解,加入三乙胺5ml,磷酸调节pH6.8)
流动相B:乙腈:甲醇—1:1
流动相:流动相A-流动相B(40:60)
柱温:35℃
流速:1.0ml/min
检测波长:320nm
进样量:5ul
分析时间:记录色谱图至主成分峰保留时间的2倍
溶剂:乙腈
(2)样品配制
供试品溶液:取4-苄氧基苯亚甲基-4-氟苯胺适量,加溶剂制成每ml含4-苄氧基苯亚甲基-4-氟苯胺3.0mg的溶液,作为供试品溶液。
对照品溶液:取SCR-5014对照品适量,加溶剂制成每ml含SCR-5014约0.6μg/ml的溶液,作为对照品溶液。
(3)测定结果
检测结果表明SCR-5014与主峰及其他杂质分离度很好。
参考:
[1] 先声药业有限公司,江苏先声药业有限公司. 一种检测4-苄氧基苯亚甲基-4-氟苯胺中的4-苄氧基苯亚甲基苯胺的HPLC方法. 2021-06-29.
[2] 南京先声东元制药有限公司,江苏先声药业有限公司. 一种检测4-苄氧基苯亚甲基-4-氟苯胺中的4-苄氧基苯亚甲基苯胺的HPLC方法. 2017-11-17.
前来学习,和同行们交流一下
前来学习,和同行们交流一下
1
引言:
镁是一种金属,属于碱土金属类元素。它以其良好的导电性和轻质特性在工业和化学领域中广泛应用。
简介:
镁主要以不溶性的碳酸盐和硫酸盐形式存在于地壳中,按重量计算,镁通常被认为是第六丰富的元素。1807年,汉弗莱·戴维爵士首次成功分离出镁。在1807至1808年间,戴维还成功分离了钠、钾、钙、钡、铍和锶。由于镁具有低密度和高强度,它常被用来与其他金属合金,广泛应用于飞机、自行车车架、车轮等结构,甚至军舰的上层建筑。此外,镁在体育用品店中也作为引火剂出售。在生物化学过程中,镁对动物体内的神经冲动传递、肌肉收缩和碳水化合物代谢等方面都起着重要作用。
1. 镁是金属还是非金属?
镁是金属、非金属还是准金属?镁是一种轻质的银白色元素金属,符号为“Mg”,原子序数为 12。镁是一种属于碱土金属的化学元素。它具有典型的金属特性,例如金属光泽、良好的导热性和导电性,以及优异的可塑性和延展性。由于镁的密度轻、强度高,因此在各个工业领域中具有重要应用,从航空航天到汽车制造等各个方面都有广泛的用途。
2. 镁的物理性质
镁具有以下物理性质:它是一种化学元素,属于金属,化学符号为Mg,位于元素周期表IIa族,原子序数为12,原子量为24.312。镁呈银白色,非常轻,相对密度为1.74,密度为1740 kg/m3。由于其重量轻且具有良好的机械强度,长期以来镁被视为工业中较为优良的结构金属。
3. 镁的化学性质
(1)镁还具有活泼的化学性质。它能够取代水中的氢气,并且通过热还原镁的盐类和氧化物可以生产多种金属。镁在室温下与水反应,虽然其反应速度比钙慢得多。钙和镁是金属还是非金属?钙是一种类似的第 2 族金属。当浸没在水中时,氢气泡会在金属表面缓慢形成;这种反应在镁粉中发生得更快。温度越高,反应发生得越快。镁与水的可逆反应可用于储存能量并运行镁基发动机。
(2)镁能够与大多数非金属及几乎所有的酸发生反应,但与大多数碱和许多有机物(如碳氢化合物、醛类、醇类、酚类、胺类、酯类和大多数油类)仅发生轻微或根本不发生反应。作为催化剂,镁可以促进多种有机反应,如缩合、还原、加成和脱卤反应。长期以来,镁通过格氏反应合成多种复杂的有机组分,这些合成反应中镁通常作为主要的催化剂。
(3)此外,镁合金通常包含铝、锰、锆、锌、稀土金属和钍等成分。
4. 为什么镁被归类为金属?
(1)镁是优良的电导体。金属中,价带与导带重叠,使得价带中的电子可以有效地参与电流传导。镁的价电子位于3s轨道,这些电子负责其电导性能。
(2)镁具有较低的电离焓。一般来说,金属的电离焓较非金属低,因为金属的价电子与原子核的结合较为松散。对于第1组和第2组金属,价电子位于s轨道,移除这些电子所需的能量较小。镁的两个价电子位于3s轨道,因此其电离焓较低。
(3)镁表现出电正性特征。金属通常通过失去价电子来达到稳定的电子配置,因此被称为电正性元素。相反,非金属通过获得电子来实现稳定,因此称为电负性元素。镁的两个价电子位于3s轨道,镁通过失去这些电子来获得稳定的电子结构,这使得它具备电正性特征。
5. 镁的独特特性
(1)镁是一种相对轻质的金属,其密度约为铝的三分之二。
(2)由于镁的熔点较低且反应性较强,在加工和应用过程中必须特别小心,以防止其腐蚀。
(3)镁具有优异的强度重量比,因此在需要减轻重量的场合非常有用。
(4)镁在机械和电气领域表现出色,因为它既是优良的热和电导体,又具有良好的阻尼特性。
(5)由于镁的高度反应性和可燃性,它广泛应用于需要高能量输出的场合,如烟花和照明弹。
(6)在医学领域,镁被用于抗酸剂和泻药等治疗用途。
6. 镁是碱性金属吗?
镁(Mg), 化学元素,元素周期表第2族(IIa)碱土金属之一,也是最轻的结构金属。
7. 镁是活性金属吗?
金属之间的主要差异在于其化学反应的活跃程度。元素周期表左下角的金属元素表现出最强的化学活性,例如锂、钠和钾能与水发生剧烈反应。随着元素在周期表中的位置向下移动,它们的反应性逐渐增强,这主要是因为这些金属的金属性增强。相对而言,第二类金属,如镁、铝、锌和锰,其化学活性较低。这些金属在室温下不会与水反应,但会迅速与酸发生反应。
镁是一种活性金属,容易与大多数非金属发生反应。镁具有很强的化学活性;它能与大多数非金属和几乎所有酸结合。
参考:
[1]https://en.wikipedia.org/wiki/Magnesium
[2]baike.baidu.com
[3]https://pubchem.ncbi.nlm.nih.gov/compound/5462224
[4]https://www.britannica.com/science/magnesium
显示全部引言:
镁是一种金属,属于碱土金属类元素。它以其良好的导电性和轻质特性在工业和化学领域中广泛应用。
简介:
镁主要以不溶性的碳酸盐和硫酸盐形式存在于地壳中,按重量计算,镁通常被认为是第六丰富的元素。1807年,汉弗莱·戴维爵士首次成功分离出镁。在1807至1808年间,戴维还成功分离了钠、钾、钙、钡、铍和锶。由于镁具有低密度和高强度,它常被用来与其他金属合金,广泛应用于飞机、自行车车架、车轮等结构,甚至军舰的上层建筑。此外,镁在体育用品店中也作为引火剂出售。在生物化学过程中,镁对动物体内的神经冲动传递、肌肉收缩和碳水化合物代谢等方面都起着重要作用。
1. 镁是金属还是非金属?
镁是金属、非金属还是准金属?镁是一种轻质的银白色元素金属,符号为“Mg”,原子序数为 12。镁是一种属于碱土金属的化学元素。它具有典型的金属特性,例如金属光泽、良好的导热性和导电性,以及优异的可塑性和延展性。由于镁的密度轻、强度高,因此在各个工业领域中具有重要应用,从航空航天到汽车制造等各个方面都有广泛的用途。
2. 镁的物理性质
镁具有以下物理性质:它是一种化学元素,属于金属,化学符号为Mg,位于元素周期表IIa族,原子序数为12,原子量为24.312。镁呈银白色,非常轻,相对密度为1.74,密度为1740 kg/m3。由于其重量轻且具有良好的机械强度,长期以来镁被视为工业中较为优良的结构金属。
3. 镁的化学性质
(1)镁还具有活泼的化学性质。它能够取代水中的氢气,并且通过热还原镁的盐类和氧化物可以生产多种金属。镁在室温下与水反应,虽然其反应速度比钙慢得多。钙和镁是金属还是非金属?钙是一种类似的第 2 族金属。当浸没在水中时,氢气泡会在金属表面缓慢形成;这种反应在镁粉中发生得更快。温度越高,反应发生得越快。镁与水的可逆反应可用于储存能量并运行镁基发动机。
(2)镁能够与大多数非金属及几乎所有的酸发生反应,但与大多数碱和许多有机物(如碳氢化合物、醛类、醇类、酚类、胺类、酯类和大多数油类)仅发生轻微或根本不发生反应。作为催化剂,镁可以促进多种有机反应,如缩合、还原、加成和脱卤反应。长期以来,镁通过格氏反应合成多种复杂的有机组分,这些合成反应中镁通常作为主要的催化剂。
(3)此外,镁合金通常包含铝、锰、锆、锌、稀土金属和钍等成分。
4. 为什么镁被归类为金属?
(1)镁是优良的电导体。金属中,价带与导带重叠,使得价带中的电子可以有效地参与电流传导。镁的价电子位于3s轨道,这些电子负责其电导性能。
(2)镁具有较低的电离焓。一般来说,金属的电离焓较非金属低,因为金属的价电子与原子核的结合较为松散。对于第1组和第2组金属,价电子位于s轨道,移除这些电子所需的能量较小。镁的两个价电子位于3s轨道,因此其电离焓较低。
(3)镁表现出电正性特征。金属通常通过失去价电子来达到稳定的电子配置,因此被称为电正性元素。相反,非金属通过获得电子来实现稳定,因此称为电负性元素。镁的两个价电子位于3s轨道,镁通过失去这些电子来获得稳定的电子结构,这使得它具备电正性特征。
5. 镁的独特特性
(1)镁是一种相对轻质的金属,其密度约为铝的三分之二。
(2)由于镁的熔点较低且反应性较强,在加工和应用过程中必须特别小心,以防止其腐蚀。
(3)镁具有优异的强度重量比,因此在需要减轻重量的场合非常有用。
(4)镁在机械和电气领域表现出色,因为它既是优良的热和电导体,又具有良好的阻尼特性。
(5)由于镁的高度反应性和可燃性,它广泛应用于需要高能量输出的场合,如烟花和照明弹。
(6)在医学领域,镁被用于抗酸剂和泻药等治疗用途。
6. 镁是碱性金属吗?
镁(Mg), 化学元素,元素周期表第2族(IIa)碱土金属之一,也是最轻的结构金属。
7. 镁是活性金属吗?
金属之间的主要差异在于其化学反应的活跃程度。元素周期表左下角的金属元素表现出最强的化学活性,例如锂、钠和钾能与水发生剧烈反应。随着元素在周期表中的位置向下移动,它们的反应性逐渐增强,这主要是因为这些金属的金属性增强。相对而言,第二类金属,如镁、铝、锌和锰,其化学活性较低。这些金属在室温下不会与水反应,但会迅速与酸发生反应。
镁是一种活性金属,容易与大多数非金属发生反应。镁具有很强的化学活性;它能与大多数非金属和几乎所有酸结合。
参考:
[1]https://en.wikipedia.org/wiki/Magnesium
[2]baike.baidu.com
[3]https://pubchem.ncbi.nlm.nih.gov/compound/5462224
[4]https://www.britannica.com/science/magnesium
1
引言:
测定维生素 K4 的含量对于保证其在药物和营养补充品中的有效性至关重要。正确的分析技术能够准确量化维生素 K4 的浓度,以确保符合相关的质量标准。
简述:
维生素K4(VK4)是一种水溶性的甲萘醌化合物,为VK2的人工合成衍生物,最初被用作临床止血药物。近些年已经有研究表明VK与血管健康、骨代谢有着密切关系,VK作为γ-亚麻酸蛋白家族如凝血素、凝血因子Ⅶ、凝血因子Ⅸ和凝血因子Ⅹ合成中的辅因子,在凝血系统中扮演了很重要的角色。
维生素 K 是一类拥有同样甲基萘醌环并且带有不同侧链的有机化合物的统称。有VK1、VK2、VK3、VK4 等几种形式,VK1(叶绿醌)是从绿色植物中提取的,VK2(甲基萘醌)是由肠道细菌(如大肠杆菌)合成的。而 VK3、VK4 是 VK1、VK2 的人工合成衍生物,属水溶性的维生素。VK2 还能根据侧链长短衍生出不同的化合物,被称为甲萘醌—MK-n,n 代表了侧链中异戊烯的个数。VK3 是一种合成的化合物,它缺少一个侧链,但因其能够在体内转化为 MK-4 而具有生物学活性。和营养学关系最密切的甲萘醌是 MK-4 以及 MK-7—MK-9。有证据表明 VK1 能够在体内转化为 MK-4。
VK 为黄色晶体,熔点为 52 ℃到 54 ℃,通常呈油状液体或固体,不溶于水,能溶于油脂及醚等有机溶剂。 所有 VK 的化学性质都较稳定,能耐酸、耐热,正常烹调中只有很少损失,对光敏感,也易被碱和紫外线分解。
膳食中 VK 都是脂溶性的,吸收需要胆汁协助,所以主要由小肠吸收进入淋巴系统,且其吸收取决于胰腺和胆囊的功能,在正常情况下其中约 40-70%可被吸收。其在人体内的半衰期比较短,约 17 小时。
检测:
1. 高效液相色谱法测定维生素K4 的含量
孙兵等人采用YWG-C18柱,以甲醇∶水=85∶15(体积比)为流动相,流速为0.5 mL/min,以285 nm为检测波长,建立维生素K4(化学名为2-甲基-1,4萘二酚双醋酸酯)的高效液相色谱法。该方法在2~10 μg范围内线性关系良好,相关系数为0.999 8.回收率99.15%~100.8%,相对标准偏差为0.66%。该方法操作简单,分析速度快,结果准确,适用于维生素K4的产品检验。
2. 化学除氧-胶束增稳室温嶙光法测定维生素K4
刘秀萍等人在Na2SO3化学除氧的十二烷基硫酸钠(SDS)胶束体系中,以T1Ac为重原子微扰剂,进行了维生素K4的室温燐光法(RTP)测定。研究发现样品温度对RTP发射强度有影响,重原子T1/SDS有一个临界比率值为40%,到此临界比率值后能获得高的RTP值。介质pH、Na2SO3、SDS及TlAc浓度不仅影响RTP强度,而且影响体系的除氧速度,最佳pH值范围在7.0~7.8之间,分析校正曲线在1×10-6mol/L~8×10-6mol/L和1×10-5mol/L~4×10-5mol/L呈良好的线性关系,方法的检出限(DL)为2.1×10-6mol/L,相对标准偏差(RSD)为4.4%。
参考:
[1] 孙兵,金丹,臧娜,等. 高效液相色谱法测定维生素K4 的含量[J]. 应用化工,2009,28(4):615-616. DOI:10.3969/j.issn.1671-3206.2009.04.037.
[2] 刘秀萍,袁宏,董川,等. 化学除氧-胶束增稳室温嶙光法测定维生素K4[J]. 山西大学学报(自然科学版),2003,26(1):43-45. DOI:10.3969/j.issn.0253-2395.2003.01.011.
[3] 姜雨. 维生素K4对人前列腺癌PC-3细胞凋亡作用的研究[D]. 辽宁:辽宁师范大学,2013. DOI:10.7666/d.Y2377104.
显示全部引言:
测定维生素 K4 的含量对于保证其在药物和营养补充品中的有效性至关重要。正确的分析技术能够准确量化维生素 K4 的浓度,以确保符合相关的质量标准。
简述:
维生素K4(VK4)是一种水溶性的甲萘醌化合物,为VK2的人工合成衍生物,最初被用作临床止血药物。近些年已经有研究表明VK与血管健康、骨代谢有着密切关系,VK作为γ-亚麻酸蛋白家族如凝血素、凝血因子Ⅶ、凝血因子Ⅸ和凝血因子Ⅹ合成中的辅因子,在凝血系统中扮演了很重要的角色。
维生素 K 是一类拥有同样甲基萘醌环并且带有不同侧链的有机化合物的统称。有VK1、VK2、VK3、VK4 等几种形式,VK1(叶绿醌)是从绿色植物中提取的,VK2(甲基萘醌)是由肠道细菌(如大肠杆菌)合成的。而 VK3、VK4 是 VK1、VK2 的人工合成衍生物,属水溶性的维生素。VK2 还能根据侧链长短衍生出不同的化合物,被称为甲萘醌—MK-n,n 代表了侧链中异戊烯的个数。VK3 是一种合成的化合物,它缺少一个侧链,但因其能够在体内转化为 MK-4 而具有生物学活性。和营养学关系最密切的甲萘醌是 MK-4 以及 MK-7—MK-9。有证据表明 VK1 能够在体内转化为 MK-4。
VK 为黄色晶体,熔点为 52 ℃到 54 ℃,通常呈油状液体或固体,不溶于水,能溶于油脂及醚等有机溶剂。 所有 VK 的化学性质都较稳定,能耐酸、耐热,正常烹调中只有很少损失,对光敏感,也易被碱和紫外线分解。
膳食中 VK 都是脂溶性的,吸收需要胆汁协助,所以主要由小肠吸收进入淋巴系统,且其吸收取决于胰腺和胆囊的功能,在正常情况下其中约 40-70%可被吸收。其在人体内的半衰期比较短,约 17 小时。
检测:
1. 高效液相色谱法测定维生素K4 的含量
孙兵等人采用YWG-C18柱,以甲醇∶水=85∶15(体积比)为流动相,流速为0.5 mL/min,以285 nm为检测波长,建立维生素K4(化学名为2-甲基-1,4萘二酚双醋酸酯)的高效液相色谱法。该方法在2~10 μg范围内线性关系良好,相关系数为0.999 8.回收率99.15%~100.8%,相对标准偏差为0.66%。该方法操作简单,分析速度快,结果准确,适用于维生素K4的产品检验。
2. 化学除氧-胶束增稳室温嶙光法测定维生素K4
刘秀萍等人在Na2SO3化学除氧的十二烷基硫酸钠(SDS)胶束体系中,以T1Ac为重原子微扰剂,进行了维生素K4的室温燐光法(RTP)测定。研究发现样品温度对RTP发射强度有影响,重原子T1/SDS有一个临界比率值为40%,到此临界比率值后能获得高的RTP值。介质pH、Na2SO3、SDS及TlAc浓度不仅影响RTP强度,而且影响体系的除氧速度,最佳pH值范围在7.0~7.8之间,分析校正曲线在1×10-6mol/L~8×10-6mol/L和1×10-5mol/L~4×10-5mol/L呈良好的线性关系,方法的检出限(DL)为2.1×10-6mol/L,相对标准偏差(RSD)为4.4%。
参考:
[1] 孙兵,金丹,臧娜,等. 高效液相色谱法测定维生素K4 的含量[J]. 应用化工,2009,28(4):615-616. DOI:10.3969/j.issn.1671-3206.2009.04.037.
[2] 刘秀萍,袁宏,董川,等. 化学除氧-胶束增稳室温嶙光法测定维生素K4[J]. 山西大学学报(自然科学版),2003,26(1):43-45. DOI:10.3969/j.issn.0253-2395.2003.01.011.
[3] 姜雨. 维生素K4对人前列腺癌PC-3细胞凋亡作用的研究[D]. 辽宁:辽宁师范大学,2013. DOI:10.7666/d.Y2377104.
1
急需海洋测绘 海洋生态学 海洋物理学 中级工程师
中级 高级建筑专业 能配合社保各要一名
广东第三方教育机构需注安化工 2名 能配合出场
环境环保中高级 贵州多家企业急需
海洋测绘 海洋生态学 海洋物理学 中级各需一名
根据工程师实际情况匹配合适企业
陈工:189-3323-9971(微同)1有8证9书3可3以2加3我9微9信7聊1
显示全部急需海洋测绘 海洋生态学 海洋物理学 中级工程师
中级 高级建筑专业 能配合社保各要一名
广东第三方教育机构需注安化工 2名 能配合出场
环境环保中高级 贵州多家企业急需
海洋测绘 海洋生态学 海洋物理学 中级各需一名
根据工程师实际情况匹配合适企业
陈工:189-3323-9971(微同)1有8证9书3可3以2加3我9微9信7聊1
1
引言:
测定盐酸益母草碱的含量是评估中药质量的重要步骤之一。盐酸益母草碱作为益母草中的主要活性成分,其含量的准确测定对于确保药品安全性和疗效具有关键意义。
简述:
益母草是传统的活血化瘀类中药,又称茺蔚、坤草、月母草、益母蒿等,具有活血调经、利尿消肿、清热解毒之功能,素有“血家圣药”、“经产良药”之称。清·陈士铎《本草新编》记载:“胎前、产后,皆可用之,去死胎最效,行瘀生新,亦能下乳。其名益母,有益于妇人不浅。”因此,几个世纪以来,被广泛用于促进血液循环、调节月经等妇科疾病。
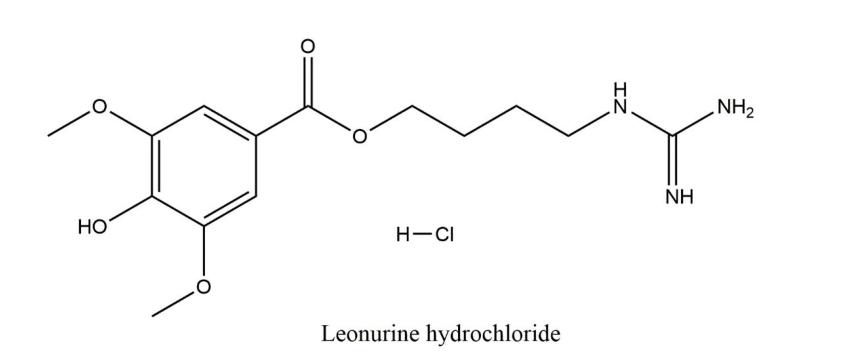
益母草碱(Leonurine)是益母草的主要活性物质之一,具有显著的直接扩张外周血管、增加血流量、抗血小板聚集等作用,可有效的降低血液黏度和提高红细胞的变形能力,还可调经止血,保护心肌缺血再灌注损伤。盐酸益母草碱的结构如下图所示:
含量测定研究:
1. 报道一
仇正英等人制备注射型盐酸益母草碱穴位埋植凝胶,建立其药物含量测定方法。以聚乳酸-羟基乙酸共聚物(PLGA)为主要凝胶载体材料,利用高效液相色谱法(HPLC),色谱柱为Eclipse Plus C18分析柱(4.6 mm×250 mm,5μm),流动相0.4%辛烷磺酸钠的0.1%磷酸溶液-乙腈(76∶24),流速1.0 mL/min,检测波长277 nm,柱温30℃,建立药物含量测定方法。通过凝胶粘度、通针阻力及体外释放度测定评价凝胶处方。
结果表明,所建立的HPLC方法,盐酸益母草碱在2.02~101μg/mL范围内线性良好;平均回收率为104.05%。注射型穴位埋植凝胶的最佳方案为3%盐酸益母草碱,20%PLGA,15%PEG400,60%甘油缩甲醛,2%HPMC,凝胶25℃粘度为193.7 mPa·s,37℃粘度为148.0 mPa·s,粘度流变为205 mPa·s;1 mL通针阻力为1.81 N,5 mL通针阻力为1.95 N;体外释放率在1 d时约达到64.50%,5 d时为91.02%。说明注射型盐酸益母草碱穴位埋植凝胶制备成功,所建立的HPLC方法可用于盐酸益母草碱埋植凝胶的质量控制。
2. 报道二
王宁莉等人建立了测定益母草颗粒中盐酸水苏碱、盐酸益母草碱含量的HPLC测定法。研究采用高效液相色谱法,以Kromasil 100-5NH2(Dimensions 250×4.6 mm,5μm)E78824和Agilent TC-C18(2)(250×4.6 mm,5μm)588925-902为色谱柱,检测波长:192 nm和277 nm,流动相:乙腈-水(80∶20)、乙腈-0.3%庚烷磺酸钠的0.1%磷酸溶液(24∶76),流速为1.0 m L/min。理论板数:按盐酸水苏碱峰计算应不低于5000、按盐酸益母草碱峰计算应不低于8000。
结果表明,盐酸水苏碱在0.181.08 mg范围内与峰面积呈良好的线性关系,r=0.99998,平均回收率为99.6%,RSD小于2.0%;盐酸益母草碱在741μg范围内与峰面积呈良好的线性关系,r=0.99998,平均回收率为99.8%,RSD小于2.0%。该方法操作简便、快速、准确、灵敏度高、重现性好,适用于益母草中盐酸水苏碱、盐酸益母草碱的含量测定。
3. 报道二
张玉萌等人建立了益母草饮片中盐酸水苏碱、盐酸益母草碱的含量测定方法,为其质量控制提供依据。研究采用高效液相色谱法对益母草饮片中盐酸水苏碱的含量进行测定,色谱柱:SCX强阳离子柱(4.6 mm×250mm,5μm);流动相:0.5 mol磷酸二氢钾-三乙胺(1 000∶1.5)(磷酸调pH值2.252.3);检测波长:192 nm;流速1.0 mL.min-1;柱温:25℃。
盐酸水苏碱的浓度在0.22.0 mg.mL-1范围内线性关系良好,RSD=1.7%(n=5);盐酸益母草碱的浓度在0.22.0 mg.mL-1范围内线性关系良好,RSD=0.08%(n=5)。该法简便、灵敏、准确,可用于益母草饮片的质量控制。
参考:
[1]廖利. 益母草及其特异性成分盐酸益母草碱的抗血栓作用及机制研究[D]. 成都中医药大学, 2022. DOI:10.26988/d.cnki.gcdzu.2022.000015.
[2]仇正英,李刚,辛蕊华,等. 注射型盐酸益母草碱穴位埋植凝胶的制备及其含量测定 [J]. 中兽医医药杂志, 2020, 39 (05): 29-34. DOI:10.13823/j.cnki.jtcvm.2020.05.007.
[3]王宁莉,张书华,王丽霞. 益母草颗粒中盐酸水苏碱与盐酸益母草碱的含量测定 [J]. 延安大学学报(医学科学版), 2015, 13 (02): 4-6+10.
[4]张玉萌,项菲菲. 益母草饮片中盐酸水苏碱、盐酸益母草碱含量测定 [J]. 辽宁中医药大学学报, 2013, 15 (02): 73-74. DOI:10.13194/j.jlunivtcm.2013.02.75.zhangym.066.
[5]旌德新星生物科技有限公司. 一种盐酸益母草碱α晶型及其制备方法和应用:CN201810047134.7[P]. 2020-12-15.
显示全部引言:
测定盐酸益母草碱的含量是评估中药质量的重要步骤之一。盐酸益母草碱作为益母草中的主要活性成分,其含量的准确测定对于确保药品安全性和疗效具有关键意义。
简述:
益母草是传统的活血化瘀类中药,又称茺蔚、坤草、月母草、益母蒿等,具有活血调经、利尿消肿、清热解毒之功能,素有“血家圣药”、“经产良药”之称。清·陈士铎《本草新编》记载:“胎前、产后,皆可用之,去死胎最效,行瘀生新,亦能下乳。其名益母,有益于妇人不浅。”因此,几个世纪以来,被广泛用于促进血液循环、调节月经等妇科疾病。
益母草碱(Leonurine)是益母草的主要活性物质之一,具有显著的直接扩张外周血管、增加血流量、抗血小板聚集等作用,可有效的降低血液黏度和提高红细胞的变形能力,还可调经止血,保护心肌缺血再灌注损伤。盐酸益母草碱的结构如下图所示:
含量测定研究:
1. 报道一
仇正英等人制备注射型盐酸益母草碱穴位埋植凝胶,建立其药物含量测定方法。以聚乳酸-羟基乙酸共聚物(PLGA)为主要凝胶载体材料,利用高效液相色谱法(HPLC),色谱柱为Eclipse Plus C18分析柱(4.6 mm×250 mm,5μm),流动相0.4%辛烷磺酸钠的0.1%磷酸溶液-乙腈(76∶24),流速1.0 mL/min,检测波长277 nm,柱温30℃,建立药物含量测定方法。通过凝胶粘度、通针阻力及体外释放度测定评价凝胶处方。
结果表明,所建立的HPLC方法,盐酸益母草碱在2.02~101μg/mL范围内线性良好;平均回收率为104.05%。注射型穴位埋植凝胶的最佳方案为3%盐酸益母草碱,20%PLGA,15%PEG400,60%甘油缩甲醛,2%HPMC,凝胶25℃粘度为193.7 mPa·s,37℃粘度为148.0 mPa·s,粘度流变为205 mPa·s;1 mL通针阻力为1.81 N,5 mL通针阻力为1.95 N;体外释放率在1 d时约达到64.50%,5 d时为91.02%。说明注射型盐酸益母草碱穴位埋植凝胶制备成功,所建立的HPLC方法可用于盐酸益母草碱埋植凝胶的质量控制。
2. 报道二
王宁莉等人建立了测定益母草颗粒中盐酸水苏碱、盐酸益母草碱含量的HPLC测定法。研究采用高效液相色谱法,以Kromasil 100-5NH2(Dimensions 250×4.6 mm,5μm)E78824和Agilent TC-C18(2)(250×4.6 mm,5μm)588925-902为色谱柱,检测波长:192 nm和277 nm,流动相:乙腈-水(80∶20)、乙腈-0.3%庚烷磺酸钠的0.1%磷酸溶液(24∶76),流速为1.0 m L/min。理论板数:按盐酸水苏碱峰计算应不低于5000、按盐酸益母草碱峰计算应不低于8000。
结果表明,盐酸水苏碱在0.181.08 mg范围内与峰面积呈良好的线性关系,r=0.99998,平均回收率为99.6%,RSD小于2.0%;盐酸益母草碱在741μg范围内与峰面积呈良好的线性关系,r=0.99998,平均回收率为99.8%,RSD小于2.0%。该方法操作简便、快速、准确、灵敏度高、重现性好,适用于益母草中盐酸水苏碱、盐酸益母草碱的含量测定。
3. 报道二
张玉萌等人建立了益母草饮片中盐酸水苏碱、盐酸益母草碱的含量测定方法,为其质量控制提供依据。研究采用高效液相色谱法对益母草饮片中盐酸水苏碱的含量进行测定,色谱柱:SCX强阳离子柱(4.6 mm×250mm,5μm);流动相:0.5 mol磷酸二氢钾-三乙胺(1 000∶1.5)(磷酸调pH值2.252.3);检测波长:192 nm;流速1.0 mL.min-1;柱温:25℃。
盐酸水苏碱的浓度在0.22.0 mg.mL-1范围内线性关系良好,RSD=1.7%(n=5);盐酸益母草碱的浓度在0.22.0 mg.mL-1范围内线性关系良好,RSD=0.08%(n=5)。该法简便、灵敏、准确,可用于益母草饮片的质量控制。
参考:
[1]廖利. 益母草及其特异性成分盐酸益母草碱的抗血栓作用及机制研究[D]. 成都中医药大学, 2022. DOI:10.26988/d.cnki.gcdzu.2022.000015.
[2]仇正英,李刚,辛蕊华,等. 注射型盐酸益母草碱穴位埋植凝胶的制备及其含量测定 [J]. 中兽医医药杂志, 2020, 39 (05): 29-34. DOI:10.13823/j.cnki.jtcvm.2020.05.007.
[3]王宁莉,张书华,王丽霞. 益母草颗粒中盐酸水苏碱与盐酸益母草碱的含量测定 [J]. 延安大学学报(医学科学版), 2015, 13 (02): 4-6+10.
[4]张玉萌,项菲菲. 益母草饮片中盐酸水苏碱、盐酸益母草碱含量测定 [J]. 辽宁中医药大学学报, 2013, 15 (02): 73-74. DOI:10.13194/j.jlunivtcm.2013.02.75.zhangym.066.
[5]旌德新星生物科技有限公司. 一种盐酸益母草碱α晶型及其制备方法和应用:CN201810047134.7[P]. 2020-12-15.
1
引言:
为了确保聚合物产品在使用过程中能够保持长久的光学性能和物理性能,使用光稳定剂是至关重要的。HS-944作为一种受阻胺光稳定剂,在聚合物材料中起着关键作用。为了确定其在材料中的浓度和分布情况,需要进行精确的测定和分析。
简介:
塑料食品包装材料中聚合物在其生产、运输、存储及使用等过程中,容易受到光、热、辐射等影响发生老化和降解,导致发生涉及物理、化学以及力学性能的一些变化,使其性能降低。所以一般在材料中会加入一定的光稳定剂,受阻胺光稳定剂就是一类防止或者延缓高分子塑料材料老化和降解的一类化合物。其中,最常见的受阻胺光稳定剂是 HS-944,它是一种新型、高性能的光稳定剂,能有效吸收紫外光,并具有一定的抗氧化性。在塑料材料中添加HS-944作为受阻胺光稳定剂,能有效减少辐射致色和钝化过渡金属离子等作用,从而延缓材料的老化和降解过程。随着高分子材料研究及应用的不断进展,新型塑料在食品包装等领域的应用日益增多。受阻胺类光稳定剂以其独特的光稳定性和强大的抗氧化能力等特性,因而在食品包装材料中的应用备受关注和推广。受阻胺光稳定剂 HS-944的结构如下:
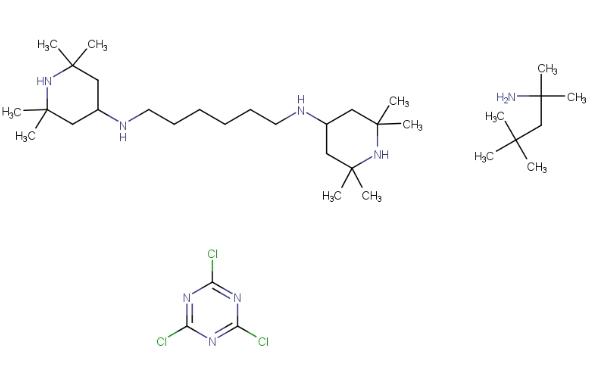
1. 性质
受阻胺光稳定剂是一种新型高性能的光稳定剂,其具有良好的抗氧化性、吸收紫外光、减小聚合物辐射致色、钝化过渡金属离子和捕捉氧等功能。高分子材料在加工、存储、使用的过程中,材料在氧、光、热的影响下容易老化和降解,导致发生物理、化学以及力学性能的变化。受阻胺光稳定剂就是一类防止或者延缓高分子材料老化和降解的一类化合物。随着高分子材料的不断发展以及相关领域研究的不断深人,受阻胺类因其结构独特,光稳定效果较传统的吸收性光稳定剂大幅提高,将会被越来越广泛地应用。
2. 测定方法
(1)报道一
黄雪琳等人采用对硝基苯甲酰氯为衍生化试剂,建立了高效液相色谱法测定食品塑料包装材料中高分子受阻胺光稳定剂HS-944的分析方法。分别通过实验优化选择色谱柱为Agilent ZORBAX SB-Aq,分析波长为400 nm,及流动相为乙腈。采用外标法定量,受阻胺光稳定剂HS-944在0.1~20 mg/L范围内具有良好的线性关系,相关系数大于0.999,检出限为10μg/L。样品加标回收率为87.28%~95.64%,相对标准偏差小于9.36%,并进行实际样品测试,结果令人满意。
(2)报道二
孙爱兵等人提出了一种针对光稳定剂受阻胺944(HS-944)新的检测方法。通过HS-944与具有紫外吸收的对硝基苯甲酰氯在四氢呋喃溶剂中发生酰胺化反应,分别对上层清夜进行紫外可见分光光度法测试实现定量分析,对反应后的沉淀物进行傅里叶红外光谱分析实现光稳定剂HS-944的定性分析。紫外可见分光光度计检测波长为254 nm。
参考:
[1] 孙爱兵,赵芳萍. 酰胺化紫外可见分光光度计法检测受阻胺[J]. 塑料工业,2018,46(3):71-73. DOI:10.3969/j.issn.1005-5770.2018.03.015.
[2] 黄雪琳,蒋小良,杨丽,等. 食品塑料包装材料中光稳定剂HS-944测定方法研究[J]. 中国口岸科学技术,2020(6):57-63. DOI:10.3969/j.issn.1002-4689.2020.06.008.
显示全部引言:
为了确保聚合物产品在使用过程中能够保持长久的光学性能和物理性能,使用光稳定剂是至关重要的。HS-944作为一种受阻胺光稳定剂,在聚合物材料中起着关键作用。为了确定其在材料中的浓度和分布情况,需要进行精确的测定和分析。
简介:
塑料食品包装材料中聚合物在其生产、运输、存储及使用等过程中,容易受到光、热、辐射等影响发生老化和降解,导致发生涉及物理、化学以及力学性能的一些变化,使其性能降低。所以一般在材料中会加入一定的光稳定剂,受阻胺光稳定剂就是一类防止或者延缓高分子塑料材料老化和降解的一类化合物。其中,最常见的受阻胺光稳定剂是 HS-944,它是一种新型、高性能的光稳定剂,能有效吸收紫外光,并具有一定的抗氧化性。在塑料材料中添加HS-944作为受阻胺光稳定剂,能有效减少辐射致色和钝化过渡金属离子等作用,从而延缓材料的老化和降解过程。随着高分子材料研究及应用的不断进展,新型塑料在食品包装等领域的应用日益增多。受阻胺类光稳定剂以其独特的光稳定性和强大的抗氧化能力等特性,因而在食品包装材料中的应用备受关注和推广。受阻胺光稳定剂 HS-944的结构如下:
1. 性质
受阻胺光稳定剂是一种新型高性能的光稳定剂,其具有良好的抗氧化性、吸收紫外光、减小聚合物辐射致色、钝化过渡金属离子和捕捉氧等功能。高分子材料在加工、存储、使用的过程中,材料在氧、光、热的影响下容易老化和降解,导致发生物理、化学以及力学性能的变化。受阻胺光稳定剂就是一类防止或者延缓高分子材料老化和降解的一类化合物。随着高分子材料的不断发展以及相关领域研究的不断深人,受阻胺类因其结构独特,光稳定效果较传统的吸收性光稳定剂大幅提高,将会被越来越广泛地应用。
2. 测定方法
(1)报道一
黄雪琳等人采用对硝基苯甲酰氯为衍生化试剂,建立了高效液相色谱法测定食品塑料包装材料中高分子受阻胺光稳定剂HS-944的分析方法。分别通过实验优化选择色谱柱为Agilent ZORBAX SB-Aq,分析波长为400 nm,及流动相为乙腈。采用外标法定量,受阻胺光稳定剂HS-944在0.1~20 mg/L范围内具有良好的线性关系,相关系数大于0.999,检出限为10μg/L。样品加标回收率为87.28%~95.64%,相对标准偏差小于9.36%,并进行实际样品测试,结果令人满意。
(2)报道二
孙爱兵等人提出了一种针对光稳定剂受阻胺944(HS-944)新的检测方法。通过HS-944与具有紫外吸收的对硝基苯甲酰氯在四氢呋喃溶剂中发生酰胺化反应,分别对上层清夜进行紫外可见分光光度法测试实现定量分析,对反应后的沉淀物进行傅里叶红外光谱分析实现光稳定剂HS-944的定性分析。紫外可见分光光度计检测波长为254 nm。
参考:
[1] 孙爱兵,赵芳萍. 酰胺化紫外可见分光光度计法检测受阻胺[J]. 塑料工业,2018,46(3):71-73. DOI:10.3969/j.issn.1005-5770.2018.03.015.
[2] 黄雪琳,蒋小良,杨丽,等. 食品塑料包装材料中光稳定剂HS-944测定方法研究[J]. 中国口岸科学技术,2020(6):57-63. DOI:10.3969/j.issn.1002-4689.2020.06.008.
1
引言:
如何对3-(1-萘基)-L-丙氨酸进行定性和定量分析是化学分析领域中的关键问题。现有多种方法,包括薄层色谱法、液相色谱法以及最新发展的LC-MS方法,为其分析提供了多样化和精确度的选择。
简述:
3-(1-萘基)-L-丙氨酸,英文名称:3-(1-Naphthyl)-L-alanine,CAS:55516-54-6,分子式:C13H13NO2,外观与性状:白色或黄色粉末,密度:1.254 g/cm3,折射率:1.678。3-(1-萘基)-L-丙氨酸是合成药物和多肽的中间体。
背景:
不同制造商生产的3-(1-萘基)-L-丙氨酸的含量各异,导致产品的纯度有显著差异,进而影响其合成品质和临床疗效。因此,准确测定3-(1-萘基)-L-丙氨酸的含量比例对于产品质量控制和确保临床疗效至关重要。已有文献报道采用薄层色谱法和液相色谱法进行其含量分析,但这些方法无法同时进行定性分析。姚颖等人提出的LC-MS方法为3-(1-萘基)-L-丙氨酸的定性和定量分析提供了一种新途径。
LC-MS分析:
1. 实验方法
1.1 样品的制备
称取3-(1-萘基)-L-丙氨酸样品(以下简称样品)适量,用流动相配置成1 mg·mL-1溶液,即用即配。
1.2 LC-MS条件
(1)色谱条件
色谱柱:ZORBAX SB-C18(2.1×150 mm 5μm);柱温:25℃;进样量1μL
流动相A为0.05%TFA溶液,流动相B为乙睛溶液:

DAD检测波长:220 nm
(2)质谱条件
电喷雾电离源(ESI),负离子检测,雾化器压力:552 kPa;干燥气温度:350℃;流速:10L/min;喷雾电压:4000 V;源内CID电压:70V;扫描方式(SIM模式)m/z:214;扫描速度:0.60 s(全程);峰宽:0.10 min。二极管阵列(DAD)与质谱串联,两者之间的滞后时间是0.2min。
2. 结果与讨论
2.1 定性分析
为了确定3-(1-萘基)-L-丙氨酸,采用二极管阵列(DAD)和负离子检测,建立了3-(1-萘基)-L-丙氨酸的分析方法(图1),其质谱采用SIM模式,在m/z为214处有较强的响应。根据其质谱特征,推测该组分为3-(1-萘基)-L-丙氨酸(图2)。
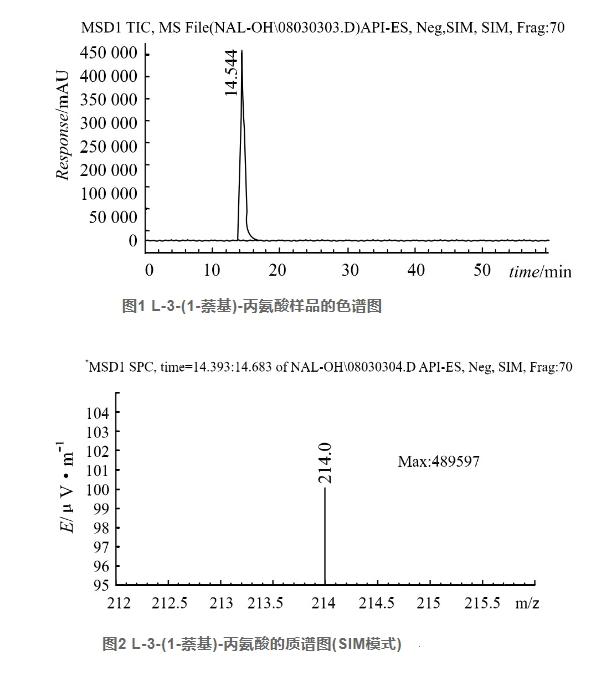
2.2 定量分析
在LC-MS方法中,采用全波长、单一波长(λ=220nm)和总离子流3种检测方式,以面积归一化法测定主峰的纯度,检测结果见表1,用3种检测方式所测定的主峰的纯度基本一致。因此,LC-MS方法可以替代液相色谱法,在进行定量的同时,可以对样品中的有关物质进行定性分析并且能够达到较低的定量下限。
2.3 结果
经过研究,该方法具有良好的专属性和较低的检测浓度(10 ng/mL,S/N=10),在整个线性范围内(10ng/mL~1.0 mg/mL)准确可靠,符合分析方法验证的要求,可以作为3-(1-萘基)-L-丙氨酸的浓度测定的方法。
参考:
[1]姚颖,谢军燕. L-3-(1-萘基)-丙氨酸的LC-MS分析[J]. 上海计量测试,2009,36(3):7-9. DOI:10.3969/j.issn.1673-2235.2009.03.003.
[2]国家药典委员会.中华人民共和国药典:3部[S].2005版.北京:化学工业出版社,2005
[3]欧洲药典.EPS:1204[S]
显示全部引言:
如何对3-(1-萘基)-L-丙氨酸进行定性和定量分析是化学分析领域中的关键问题。现有多种方法,包括薄层色谱法、液相色谱法以及最新发展的LC-MS方法,为其分析提供了多样化和精确度的选择。
简述:
3-(1-萘基)-L-丙氨酸,英文名称:3-(1-Naphthyl)-L-alanine,CAS:55516-54-6,分子式:C13H13NO2,外观与性状:白色或黄色粉末,密度:1.254 g/cm3,折射率:1.678。3-(1-萘基)-L-丙氨酸是合成药物和多肽的中间体。
背景:
不同制造商生产的3-(1-萘基)-L-丙氨酸的含量各异,导致产品的纯度有显著差异,进而影响其合成品质和临床疗效。因此,准确测定3-(1-萘基)-L-丙氨酸的含量比例对于产品质量控制和确保临床疗效至关重要。已有文献报道采用薄层色谱法和液相色谱法进行其含量分析,但这些方法无法同时进行定性分析。姚颖等人提出的LC-MS方法为3-(1-萘基)-L-丙氨酸的定性和定量分析提供了一种新途径。
LC-MS分析:
1. 实验方法
1.1 样品的制备
称取3-(1-萘基)-L-丙氨酸样品(以下简称样品)适量,用流动相配置成1 mg·mL-1溶液,即用即配。
1.2 LC-MS条件
(1)色谱条件
色谱柱:ZORBAX SB-C18(2.1×150 mm 5μm);柱温:25℃;进样量1μL
流动相A为0.05%TFA溶液,流动相B为乙睛溶液:
DAD检测波长:220 nm
(2)质谱条件
电喷雾电离源(ESI),负离子检测,雾化器压力:552 kPa;干燥气温度:350℃;流速:10L/min;喷雾电压:4000 V;源内CID电压:70V;扫描方式(SIM模式)m/z:214;扫描速度:0.60 s(全程);峰宽:0.10 min。二极管阵列(DAD)与质谱串联,两者之间的滞后时间是0.2min。
2. 结果与讨论
2.1 定性分析
为了确定3-(1-萘基)-L-丙氨酸,采用二极管阵列(DAD)和负离子检测,建立了3-(1-萘基)-L-丙氨酸的分析方法(图1),其质谱采用SIM模式,在m/z为214处有较强的响应。根据其质谱特征,推测该组分为3-(1-萘基)-L-丙氨酸(图2)。
2.2 定量分析
在LC-MS方法中,采用全波长、单一波长(λ=220nm)和总离子流3种检测方式,以面积归一化法测定主峰的纯度,检测结果见表1,用3种检测方式所测定的主峰的纯度基本一致。因此,LC-MS方法可以替代液相色谱法,在进行定量的同时,可以对样品中的有关物质进行定性分析并且能够达到较低的定量下限。
2.3 结果
经过研究,该方法具有良好的专属性和较低的检测浓度(10 ng/mL,S/N=10),在整个线性范围内(10ng/mL~1.0 mg/mL)准确可靠,符合分析方法验证的要求,可以作为3-(1-萘基)-L-丙氨酸的浓度测定的方法。
参考:
[1]姚颖,谢军燕. L-3-(1-萘基)-丙氨酸的LC-MS分析[J]. 上海计量测试,2009,36(3):7-9. DOI:10.3969/j.issn.1673-2235.2009.03.003.
[2]国家药典委员会.中华人民共和国药典:3部[S].2005版.北京:化学工业出版社,2005
[3]欧洲药典.EPS:1204[S]
1
找注安师(交通运输)+公路相关高级
不用出场 社保不限 地区不限 签两年一次性付,有意向了解欢迎随时咨询
另我司也长期收各类职称 注册 技能证,兼职全职均可根据情况匹配合适单位
陈工:189-3323-9971(微同)1有8证9书3可3以2加3我9微9信7聊1
显示全部找注安师(交通运输)+公路相关高级
不用出场 社保不限 地区不限 签两年一次性付,有意向了解欢迎随时咨询
另我司也长期收各类职称 注册 技能证,兼职全职均可根据情况匹配合适单位
陈工:189-3323-9971(微同)1有8证9书3可3以2加3我9微9信7聊1
1
引言:
通过有效的分析方法和技术手段,可以准确测定异绿原酸B的含量及其在不同物质中的分布情况,为其应用和研究提供科学依据。
简介:
金银花(学名LonicerajaponicaThunb.)是忍冬科植物,其干燥花蕾或初开的花被广泛应用于我国中药领域。它以清热解毒、凉散风热为主要功效,常用于治疗热血毒痢、风热感冒、痈肿疔疮以及瘟病发热等疾病。金银花中富含多种有机酸类化合物,具有抗菌、抗炎、抑制血小板聚集、抗血栓和抗氧化等药理活性。
异绿原酸A、B和C是二咖啡酰奎宁酸类化合物,是金银花中的有效活性成分,是一类由奎宁酸与数目不等的咖啡酸通过酯化反应缩合而成的有机酸类天然成分,广泛存在于植物界中。近20年来国内外学者就二咖啡酰奎宁酸类的植物化学和药理进行了深入研究,发现其具有一些重要生物活性,极具临床应用价值。主要的药理活性有抗氧化、抑制氧酶、抗动脉粥样硬化、抗动脉粥样硬化、抗血小板活性物质、调血脂、抗炎、抗病毒、抑制组胺释放、抗纤维化、抑制平滑肌收缩等作用。异绿原酸B的结构如下:

1. 应用
异绿原酸 B(isochlorogenic acid B,ICAB)的结构类似物绿原酸(3-咖啡酰奎尼酸)具有广泛的药理学活性。绿原酸可通过上调 Bcl-2 的表达及下调 caspase-3 的表达抑制乙醇引起的 PC12 细胞的凋亡;通过抗氧化和抗炎作用抑制 D-半乳糖引起的小鼠肝和肾损伤;通过下调 STAT3 和 NF-κB 表达,减轻 α-异硫氰酸萘酯诱导的胆汁淤积性小鼠肝损伤;也可通过增加肝细胞中 ATP 的产生,刺激线粒体氧化磷酸化和抑制糖酵解来抑制脂多糖诱导的肝损伤。此外,绿原酸还可减轻糖尿病性肾病大鼠肾组织的氧化应激损伤。而由于 ICAB 与绿原酸相比具有更多的酚羟基,被证实比绿原酸具有更好的抗氧化和抗神经退行性疾病的药理作用。
2. 含量测定研究
(1)报道一
蔡小兵等人采用UPLC法对酒续断配方颗粒的6个含量指标马钱苷酸、川续断皂苷Ⅵ、绿原酸、异绿原酸A、异绿原酸B、异绿原酸C进行含量测定,并应用一测多评法同时测定6个指标的含量,为其质量控制提供依据。
研究采用超高效液相UPLC-UV,以ACQUITYHSST3(2.1 mm×100 mm,1.8μm)为色谱柱,以乙腈-0.05%磷酸为流动相进行梯度洗脱,检测波长为220 nm,流速0.3 ml/min,进样体积2μl,柱温30℃。同时测定酒续断配方颗粒中马钱苷酸、川续断皂苷Ⅵ、绿原酸、异绿原酸A、异绿原酸B、异绿原酸C 6种成分的含量,采用一测多评法,以绿原酸为内参物,通过相对校正因子测定马钱苷酸、川续断皂苷Ⅵ、异绿原酸A、异绿原酸B、异绿原酸C含量,同时采用外标法测定多批酒续断配方颗粒中这6个指标成分的含量,比较计算值与实测值的差异,验证一测多评法的准确性。
建立相对校正因子重现性良好,采用相对校正因子计算的含量值与实测值之间无明显差异。
(2)报道二
许佩勤等人建立了一种高效液相色谱-串联质谱法同时快速、准确测定饮料中7种绿原酸类物质含量的方法。研究不同柱温、流速、色谱柱、提取溶剂、超声时间等对7种绿原酸类物质测定的影响,最终确定试样经90%甲醇水溶液超声提取,采用RP18(2.1 mm×100 mm,1.7μm)色谱柱分离,HPLC-MS/MS同时测定新绿原酸、绿原酸、隐绿原酸、异绿原酸A、异绿原酸B、异绿原酸C、1,5-二咖啡酰奎宁酸成分含量。
该检测方法简便、准确、可靠,线性关系、精密度、重复性、稳定性、加样回收率均符合要求,可用于饮料中7种绿原酸类成分的定量分析。
(3)报道三
任荣军等人建立同时测定艾绒中4种成分含量的高效液相色谱(HPLC)法。研究方法为:色谱柱为Phenomenex Luna C18(2)100A柱(250 mm×4.6 mm,5μm),流动相为乙腈-0.4%磷酸水溶液(梯度洗脱),流速为1.0 mL/min,检测波长为325 nm,柱温为30℃,进样量为10μL。
绿原酸、异绿原酸A、异绿原酸B、异绿原酸C进样量分别在39.16~391.64 ng(r=0.999 1)、41.08~410.82 ng(r=0.999 7)、46.49~464.91 ng(r=0.999 7)、47.12~471.18 ng(r=0.999 9)范围内与峰面积线性关系良好;精密度、稳定性、重复性试验结果的RSD均小于2.0%;平均加样回收率分别为100.06%,99.90%,100.12%,100.19%,RSD分别为2.31%,2.65%,2.01%,1.29%(n=6)。该方法操作简便,结果准确,重复性好,可用于艾绒的质量控制。
参考:
[1]蔡小兵,江斌,李璐,等. 一测多评法同时测定酒续断配方颗粒中6种成分的含量 [J]. 中国处方药, 2024, 22 (03): 50-55.
[2]许佩勤,陈茹,利通,等. 高效液相色谱-串联质谱法测定饮料中7种绿原酸类物质 [J]. 食品科技, 2024, 49 (02): 287-293. DOI:10.13684/j.cnki.spkj.2024.02.032.
[3]任荣军,湛建峰,陈虹球,等. 高效液相色谱法同时测定艾绒中4种成分 [J]. 中国药业, 2023, 32 (24): 103-106.
[4]倪付勇,宋亚玲,刘露,等. 异绿原酸A、B和C的制备工艺研究[J]. 中草药,2015,46(3):369-373. DOI:10.7501/j.issn.0253-2670.2015.03.012.
[5]刘鑫,徐小薇. 异绿原酸B对四氯化碳致小鼠急性肝损伤的保护作用[J]. 国际药学研究杂志,2017,44(6):531-536. DOI:10.13220/j.cnki.jipr.2017.06.013.
显示全部引言:
通过有效的分析方法和技术手段,可以准确测定异绿原酸B的含量及其在不同物质中的分布情况,为其应用和研究提供科学依据。
简介:
金银花(学名LonicerajaponicaThunb.)是忍冬科植物,其干燥花蕾或初开的花被广泛应用于我国中药领域。它以清热解毒、凉散风热为主要功效,常用于治疗热血毒痢、风热感冒、痈肿疔疮以及瘟病发热等疾病。金银花中富含多种有机酸类化合物,具有抗菌、抗炎、抑制血小板聚集、抗血栓和抗氧化等药理活性。
异绿原酸A、B和C是二咖啡酰奎宁酸类化合物,是金银花中的有效活性成分,是一类由奎宁酸与数目不等的咖啡酸通过酯化反应缩合而成的有机酸类天然成分,广泛存在于植物界中。近20年来国内外学者就二咖啡酰奎宁酸类的植物化学和药理进行了深入研究,发现其具有一些重要生物活性,极具临床应用价值。主要的药理活性有抗氧化、抑制氧酶、抗动脉粥样硬化、抗动脉粥样硬化、抗血小板活性物质、调血脂、抗炎、抗病毒、抑制组胺释放、抗纤维化、抑制平滑肌收缩等作用。异绿原酸B的结构如下:
1. 应用
异绿原酸 B(isochlorogenic acid B,ICAB)的结构类似物绿原酸(3-咖啡酰奎尼酸)具有广泛的药理学活性。绿原酸可通过上调 Bcl-2 的表达及下调 caspase-3 的表达抑制乙醇引起的 PC12 细胞的凋亡;通过抗氧化和抗炎作用抑制 D-半乳糖引起的小鼠肝和肾损伤;通过下调 STAT3 和 NF-κB 表达,减轻 α-异硫氰酸萘酯诱导的胆汁淤积性小鼠肝损伤;也可通过增加肝细胞中 ATP 的产生,刺激线粒体氧化磷酸化和抑制糖酵解来抑制脂多糖诱导的肝损伤。此外,绿原酸还可减轻糖尿病性肾病大鼠肾组织的氧化应激损伤。而由于 ICAB 与绿原酸相比具有更多的酚羟基,被证实比绿原酸具有更好的抗氧化和抗神经退行性疾病的药理作用。
2. 含量测定研究
(1)报道一
蔡小兵等人采用UPLC法对酒续断配方颗粒的6个含量指标马钱苷酸、川续断皂苷Ⅵ、绿原酸、异绿原酸A、异绿原酸B、异绿原酸C进行含量测定,并应用一测多评法同时测定6个指标的含量,为其质量控制提供依据。
研究采用超高效液相UPLC-UV,以ACQUITYHSST3(2.1 mm×100 mm,1.8μm)为色谱柱,以乙腈-0.05%磷酸为流动相进行梯度洗脱,检测波长为220 nm,流速0.3 ml/min,进样体积2μl,柱温30℃。同时测定酒续断配方颗粒中马钱苷酸、川续断皂苷Ⅵ、绿原酸、异绿原酸A、异绿原酸B、异绿原酸C 6种成分的含量,采用一测多评法,以绿原酸为内参物,通过相对校正因子测定马钱苷酸、川续断皂苷Ⅵ、异绿原酸A、异绿原酸B、异绿原酸C含量,同时采用外标法测定多批酒续断配方颗粒中这6个指标成分的含量,比较计算值与实测值的差异,验证一测多评法的准确性。
建立相对校正因子重现性良好,采用相对校正因子计算的含量值与实测值之间无明显差异。
(2)报道二
许佩勤等人建立了一种高效液相色谱-串联质谱法同时快速、准确测定饮料中7种绿原酸类物质含量的方法。研究不同柱温、流速、色谱柱、提取溶剂、超声时间等对7种绿原酸类物质测定的影响,最终确定试样经90%甲醇水溶液超声提取,采用RP18(2.1 mm×100 mm,1.7μm)色谱柱分离,HPLC-MS/MS同时测定新绿原酸、绿原酸、隐绿原酸、异绿原酸A、异绿原酸B、异绿原酸C、1,5-二咖啡酰奎宁酸成分含量。
该检测方法简便、准确、可靠,线性关系、精密度、重复性、稳定性、加样回收率均符合要求,可用于饮料中7种绿原酸类成分的定量分析。
(3)报道三
任荣军等人建立同时测定艾绒中4种成分含量的高效液相色谱(HPLC)法。研究方法为:色谱柱为Phenomenex Luna C18(2)100A柱(250 mm×4.6 mm,5μm),流动相为乙腈-0.4%磷酸水溶液(梯度洗脱),流速为1.0 mL/min,检测波长为325 nm,柱温为30℃,进样量为10μL。
绿原酸、异绿原酸A、异绿原酸B、异绿原酸C进样量分别在39.16~391.64 ng(r=0.999 1)、41.08~410.82 ng(r=0.999 7)、46.49~464.91 ng(r=0.999 7)、47.12~471.18 ng(r=0.999 9)范围内与峰面积线性关系良好;精密度、稳定性、重复性试验结果的RSD均小于2.0%;平均加样回收率分别为100.06%,99.90%,100.12%,100.19%,RSD分别为2.31%,2.65%,2.01%,1.29%(n=6)。该方法操作简便,结果准确,重复性好,可用于艾绒的质量控制。
参考:
[1]蔡小兵,江斌,李璐,等. 一测多评法同时测定酒续断配方颗粒中6种成分的含量 [J]. 中国处方药, 2024, 22 (03): 50-55.
[2]许佩勤,陈茹,利通,等. 高效液相色谱-串联质谱法测定饮料中7种绿原酸类物质 [J]. 食品科技, 2024, 49 (02): 287-293. DOI:10.13684/j.cnki.spkj.2024.02.032.
[3]任荣军,湛建峰,陈虹球,等. 高效液相色谱法同时测定艾绒中4种成分 [J]. 中国药业, 2023, 32 (24): 103-106.
[4]倪付勇,宋亚玲,刘露,等. 异绿原酸A、B和C的制备工艺研究[J]. 中草药,2015,46(3):369-373. DOI:10.7501/j.issn.0253-2670.2015.03.012.
[5]刘鑫,徐小薇. 异绿原酸B对四氯化碳致小鼠急性肝损伤的保护作用[J]. 国际药学研究杂志,2017,44(6):531-536. DOI:10.13220/j.cnki.jipr.2017.06.013.
1
引言:
如何合成并分析异氰酸异丙酯是当前有机化学研究的重要课题,通过优化合成路线和精密分析技术,可以有效地获取和评估异氰酸异丙酯的结构及其应用潜力。
简介:
异丙基异氰酸酯,又名异氰酸异丙酯,为无色至浅黄色液体,沸点74-75℃,有明显不愉快气味。异氰酸异丙酯是一种重要的有机合成中间体,尤其是合成杀菌剂异菌脲的关键中间体,并在医药化学品、农用化学品、有机合成、高分子材料合成等工业中有着广泛的应用。
1. 合成
目前异氰酸酯在工业上主要仍以光气法为主,冷热光气化法合成产率较低,液相成盐光气化法可在温和的条件下制得异氰酸酯,同时反应收率较高,但缺点是反应时间长、所需溶剂量大、副产物多且难于分离,近年来随着人类环保意识的加强,科技的不断进步,越来越多的对环境友好的绿色合成法相继出现。基于上述原因,骆建等人采用高温气相无溶剂光气法合成异丙基异氰酸酯,再引入催化剂进行脱酸,然后采用高温精馏的方法得到含量大于99%,游离氯小于0.2%异丙基异氰酸酯,从而满足市场需求,该方法具有后处理简单,反应收率高、能耗低等优点,适合工业化生产。具体如下:
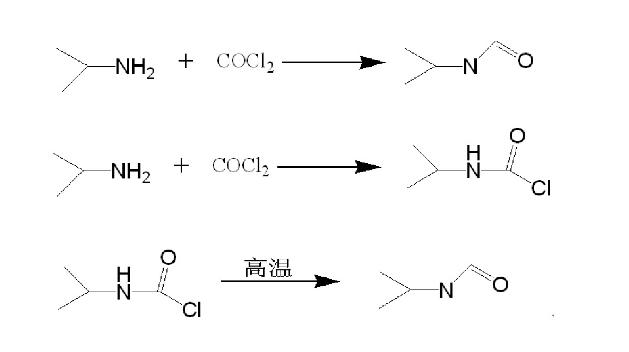
实验操作为:将预热至200℃的异丙胺、光气按异丙胺:光气=1:1.2(mol)通入管式反应器上端,管式反应器下端导入一个装有搅拌器,温度计,盘管回流冷凝器,导气管并加入0.1mol DMAP的1000 ml四口烧瓶中,开启搅拌,同时慢慢地从反应瓶底部导入氮气,赶走多余的光气和氯化氢气体,反应结束后,将反应液移入精馏塔进行精馏,控制精馏柱温度130℃以上,收集馏分,经气相色谱面积归一法分析:产品含量99。38%,化学分析法分析:游离氯小于0.15%,收率大于88.4%。
2. 分析
有研究用聚乙二醇 20000为固定液 ,醋酸丁酯为内标物,用具有氢火焰离子化检测器的气相色谱仪对异氰酸异丙酯进行定量分析,方法标准偏差为 0.10 % ,平均回收率为 100.3 %。具体如下:
2.1 气相色谱操作条件
色谱柱:20 m×4 mm (id) 玻璃毛细管柱内装聚乙二醇20000固定液;柱温:110℃,气化检测室140℃;气体流量:载气 (N2) 45 mL/min,氢气40 mL/min,空气450 mL/min。在上述操作条件下,异氰酸异丙酯和内标物的保留时间分别约为3.37 min和4.36 min,见图。
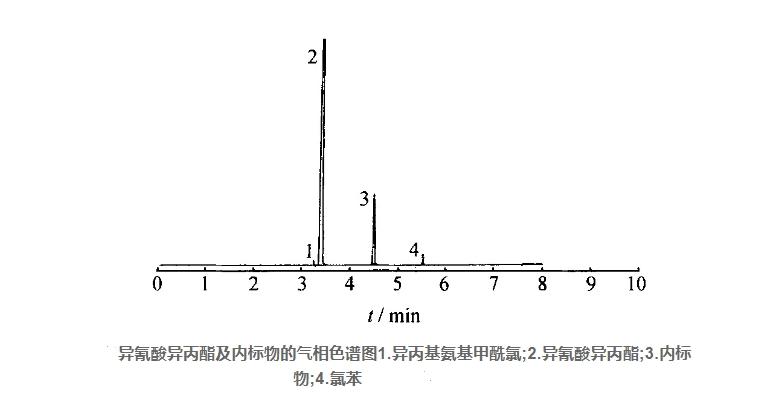
2.2 测定步骤
(1)标样溶液的配制
称取约0.25 g异氰酸异丙酯标准品 (精确到0.0001 g) ,置于具磨口塞玻璃瓶中,用移液管准确加入5 mL内标溶液,摇匀,冰浴保存。
(2)试样溶液的配制
称取约2.5 g异氰酸异丙酯粗品 (精确到0.0001 g) ,置于具磨口塞玻璃瓶中,用移液管准确加入5 mL内标溶液,摇匀,冰浴保存。
(3)试样的测定
在上述色谱操作条件下,待仪器基线稳定后,按一针标样溶液,二针试样溶液的顺序进行色谱分析。
2.3 结果
该方法简便快速且能准确测定异氰酸异丙酯,也可适用于其它脂肪族异氰酸酯的含量分析。
参考:
[1]魏星. 异氰酸异丙酯连续化生产工艺技术 [J]. 化学工程与装备, 2022, (01): 19-20. DOI:10.19566/j.cnki.cn35-1285/tq.2022.01.055.
[2]骆建. 无溶剂法合成异氰酸异丙酯 [J]. 化学工程与装备, 2021, (12): 50-51. DOI:10.19566/j.cnki.cn35-1285/tq.2021.12.021.
[3]陈凤英. 异氰酸异丙酯的气相色谱分析 [J]. 杭州师范学院学报(自然科学版), 2003, (02): 20-22.
显示全部引言:
如何合成并分析异氰酸异丙酯是当前有机化学研究的重要课题,通过优化合成路线和精密分析技术,可以有效地获取和评估异氰酸异丙酯的结构及其应用潜力。
简介:
异丙基异氰酸酯,又名异氰酸异丙酯,为无色至浅黄色液体,沸点74-75℃,有明显不愉快气味。异氰酸异丙酯是一种重要的有机合成中间体,尤其是合成杀菌剂异菌脲的关键中间体,并在医药化学品、农用化学品、有机合成、高分子材料合成等工业中有着广泛的应用。
1. 合成
目前异氰酸酯在工业上主要仍以光气法为主,冷热光气化法合成产率较低,液相成盐光气化法可在温和的条件下制得异氰酸酯,同时反应收率较高,但缺点是反应时间长、所需溶剂量大、副产物多且难于分离,近年来随着人类环保意识的加强,科技的不断进步,越来越多的对环境友好的绿色合成法相继出现。基于上述原因,骆建等人采用高温气相无溶剂光气法合成异丙基异氰酸酯,再引入催化剂进行脱酸,然后采用高温精馏的方法得到含量大于99%,游离氯小于0.2%异丙基异氰酸酯,从而满足市场需求,该方法具有后处理简单,反应收率高、能耗低等优点,适合工业化生产。具体如下:
实验操作为:将预热至200℃的异丙胺、光气按异丙胺:光气=1:1.2(mol)通入管式反应器上端,管式反应器下端导入一个装有搅拌器,温度计,盘管回流冷凝器,导气管并加入0.1mol DMAP的1000 ml四口烧瓶中,开启搅拌,同时慢慢地从反应瓶底部导入氮气,赶走多余的光气和氯化氢气体,反应结束后,将反应液移入精馏塔进行精馏,控制精馏柱温度130℃以上,收集馏分,经气相色谱面积归一法分析:产品含量99。38%,化学分析法分析:游离氯小于0.15%,收率大于88.4%。
2. 分析
有研究用聚乙二醇 20000为固定液 ,醋酸丁酯为内标物,用具有氢火焰离子化检测器的气相色谱仪对异氰酸异丙酯进行定量分析,方法标准偏差为 0.10 % ,平均回收率为 100.3 %。具体如下:
2.1 气相色谱操作条件
色谱柱:20 m×4 mm (id) 玻璃毛细管柱内装聚乙二醇20000固定液;柱温:110℃,气化检测室140℃;气体流量:载气 (N2) 45 mL/min,氢气40 mL/min,空气450 mL/min。在上述操作条件下,异氰酸异丙酯和内标物的保留时间分别约为3.37 min和4.36 min,见图。
2.2 测定步骤
(1)标样溶液的配制
称取约0.25 g异氰酸异丙酯标准品 (精确到0.0001 g) ,置于具磨口塞玻璃瓶中,用移液管准确加入5 mL内标溶液,摇匀,冰浴保存。
(2)试样溶液的配制
称取约2.5 g异氰酸异丙酯粗品 (精确到0.0001 g) ,置于具磨口塞玻璃瓶中,用移液管准确加入5 mL内标溶液,摇匀,冰浴保存。
(3)试样的测定
在上述色谱操作条件下,待仪器基线稳定后,按一针标样溶液,二针试样溶液的顺序进行色谱分析。
2.3 结果
该方法简便快速且能准确测定异氰酸异丙酯,也可适用于其它脂肪族异氰酸酯的含量分析。
参考:
[1]魏星. 异氰酸异丙酯连续化生产工艺技术 [J]. 化学工程与装备, 2022, (01): 19-20. DOI:10.19566/j.cnki.cn35-1285/tq.2022.01.055.
[2]骆建. 无溶剂法合成异氰酸异丙酯 [J]. 化学工程与装备, 2021, (12): 50-51. DOI:10.19566/j.cnki.cn35-1285/tq.2021.12.021.
[3]陈凤英. 异氰酸异丙酯的气相色谱分析 [J]. 杭州师范学院学报(自然科学版), 2003, (02): 20-22.
1
鸟苷-5'-三磷酸二钠盐的检测是化学分析研究中至关重要的一环。通过采用现代化的分析技术和方法,可以有效地检测鸟苷-5'-三磷酸二钠盐的存在及其浓度。
简述:鸟苷-5'-三磷酸二钠盐(Guanosine 5'-triphosphate,GTP)是一种嘌呤核苷三磷酸。它是转录过程中 RNA 合成所需的构建块之一。其结构类似于鸟苷核苷,唯一的区别在于像 GTP 这样的核苷酸在其核糖上具有磷酸盐。GTP 的鸟嘌呤核碱基附着在核糖的 1' 碳上,三磷酸盐部分附着在核糖的 5' 碳上。它还具有能量来源或代谢反应中底物激活剂的作用,就像 ATP 一样,但更具体。它被用作蛋白质合成和糖异生的能量来源。GTP 对信号转导至关重要,特别是对于 G 蛋白,在第二信使机制中,它通过 GTPases 的作用转化为鸟苷二磷酸 (GDP)。
鸟苷-5'-三磷酸二钠盐作为含高能量基质和多种酶的激活因子,对神经系统、代谢系统和心血管等有着非常重要的作用。GTP可以调节Ca2+诱导的胰岛素分泌],还可以刺激神经轴突的生长等。虽然GTP在生理过程中具有很重要的作用,但由于其它三磷酸物质的共存干扰,使得用简单的方法区分GTP与其结构相似物(如三磷酸腺苷(ATP)、三磷酸胞苷(CTP)、三磷酸尿苷(UTP)、二磷酸腺苷(ADP)和一磷酸腺苷(AMP))仍然是一个难题。
检测:
1. 方法一
(1)量子点的合成
将288毫克的硫酸锌七水合物和10毫克的乙酸锰(II)溶解在50毫升微波超声波反应瓶(异口瓶)中,随后加入750毫克预先溶解在少量水中的聚乙烯亚胺(PEI),并最终用水将溶液的体积调整至约20毫升。通过加入6 mol/L盐酸来调节溶液的pH值至4.0,在微波超声波合成/萃取仪中设置参数:目标温度为60摄氏度,微波功率为500瓦,超声波功率为1,000瓦,反应时间为15分钟。整个过程在氩气氛围下进行。随后加入2.5 mL 0.4 mol/L的Na2S·9H2O水溶液,反应30 min,得到PEI包覆的Mn掺杂ZnS量子点。在不加PEI的条件下重复上述操作,得到裸的Mn掺杂ZnS量子点作为对照。分别将所得的PEI包覆和裸的Mn掺杂ZnS量子点按体积比1 ∶1加入无水乙醇使量子点沉降,10 000 r/min离心10 min,倾去上层清液,所得固体放入真空干燥箱中干燥24 h,得到实验所用的PEI包覆和裸的Mn掺杂ZnS量子点。
(2)室温磷光检测
在10毫升的比色管中,首先加入1毫升pH 7.2的0.1 mol/L羟乙基哌嗪乙硫磺酸(HEPES)缓冲溶液,然后加入20微升20克/升PEI包覆的锰掺杂氧化锌硫量子点水溶液。将溶液用水稀释至10毫升定容,并充分摇匀。随后逐个加入不同浓度的GTP水溶液,在静置3分钟后使用激发波长为312纳米检测溶液的磷光。
(3)癌细胞提取液的制备
肝癌细胞中鸟苷-5'-三磷酸二钠盐按以下步骤提取:(1)HepG2细胞的复苏。从液氮罐中取出冻存管,迅速放入37 ℃水浴中使其溶解,加入一定量的培养液,800 r/min离心3 min,弃去上清液,加入培养液4 mL,吹打使细胞悬浮。重复上述步骤3次,然后将含细胞的培养液移入培养瓶中。细胞密度控制在约5×104个/mL,放入37 ℃ CO2培养箱中培养;(2)HepG2细胞的培养。培养24 h后更换培养基,如果镜检观察发现细胞数量很多,则进行传代;(3)HepG2细胞提取液的制备。首先选择体外指数生长的细胞,消化后用血球计进行计数,总量为2.5×107个/mL,然后以800 r/min离心3 min,并用预冷的PBS洗3次,800 r/min离心3 min,重悬于0.25 mL预冷的超纯水中,在0 ℃超声降解20 min使细胞壁破裂。为去除溶解产物中的细胞碎片和蛋白,依次用Amicon Ultra-4离心膜30 kDa和3 kDa离心分离(10 000 r/min×15 min)。将得到的癌细胞提取液准确稀释20倍用于检测。
2. 方法二
脱氧核酶具有独特的催化和结构性质,其中一种具有自身磷酸化能力的脱氧核酶DK2,在锰(II)存在下可将GTP上的一个磷酸基转移到5′端,而λ外切酶(λexo)可催化5′端磷酸化的双链DNA分子从5′逐渐水解到3′,但不能裂解5′-OH端。荧光染料SYBR Green I (SG I)能与双链DNA结合并产生强荧光,但与单链DNA混合时只能发出微弱的荧光。Chengzhen Hu提出了一种基于脱氧核酶DK2自身磷酸化和λexo特异性水解的新型GTP非标记荧光检测方法。该方法具有操作简便、灵敏度高、特异性好、成本低、无需荧光团(猝灭基团)标记等优点,在生物应用中具有巨大的潜力。

参考:
[1]任呼博,杨成雄,严秀平.微波-超声波辅助合成聚乙烯亚胺包覆Mn掺杂ZnS量子点用于室温磷光检测三磷酸鸟苷[J].分析测试学报,2012,31(09):1042-1050.
[2]Hu C, Jiang K, Shao Z, et al. A DNAzyme-based label-free fluorescent probe for guanosine-5′-triphosphate detection[J]. Analyst, 2020, 145(21): 6948-6954.
显示全部鸟苷-5'-三磷酸二钠盐的检测是化学分析研究中至关重要的一环。通过采用现代化的分析技术和方法,可以有效地检测鸟苷-5'-三磷酸二钠盐的存在及其浓度。
简述:鸟苷-5'-三磷酸二钠盐(Guanosine 5'-triphosphate,GTP)是一种嘌呤核苷三磷酸。它是转录过程中 RNA 合成所需的构建块之一。其结构类似于鸟苷核苷,唯一的区别在于像 GTP 这样的核苷酸在其核糖上具有磷酸盐。GTP 的鸟嘌呤核碱基附着在核糖的 1' 碳上,三磷酸盐部分附着在核糖的 5' 碳上。它还具有能量来源或代谢反应中底物激活剂的作用,就像 ATP 一样,但更具体。它被用作蛋白质合成和糖异生的能量来源。GTP 对信号转导至关重要,特别是对于 G 蛋白,在第二信使机制中,它通过 GTPases 的作用转化为鸟苷二磷酸 (GDP)。
鸟苷-5'-三磷酸二钠盐作为含高能量基质和多种酶的激活因子,对神经系统、代谢系统和心血管等有着非常重要的作用。GTP可以调节Ca2+诱导的胰岛素分泌],还可以刺激神经轴突的生长等。虽然GTP在生理过程中具有很重要的作用,但由于其它三磷酸物质的共存干扰,使得用简单的方法区分GTP与其结构相似物(如三磷酸腺苷(ATP)、三磷酸胞苷(CTP)、三磷酸尿苷(UTP)、二磷酸腺苷(ADP)和一磷酸腺苷(AMP))仍然是一个难题。
检测:
1. 方法一
(1)量子点的合成
将288毫克的硫酸锌七水合物和10毫克的乙酸锰(II)溶解在50毫升微波超声波反应瓶(异口瓶)中,随后加入750毫克预先溶解在少量水中的聚乙烯亚胺(PEI),并最终用水将溶液的体积调整至约20毫升。通过加入6 mol/L盐酸来调节溶液的pH值至4.0,在微波超声波合成/萃取仪中设置参数:目标温度为60摄氏度,微波功率为500瓦,超声波功率为1,000瓦,反应时间为15分钟。整个过程在氩气氛围下进行。随后加入2.5 mL 0.4 mol/L的Na2S·9H2O水溶液,反应30 min,得到PEI包覆的Mn掺杂ZnS量子点。在不加PEI的条件下重复上述操作,得到裸的Mn掺杂ZnS量子点作为对照。分别将所得的PEI包覆和裸的Mn掺杂ZnS量子点按体积比1 ∶1加入无水乙醇使量子点沉降,10 000 r/min离心10 min,倾去上层清液,所得固体放入真空干燥箱中干燥24 h,得到实验所用的PEI包覆和裸的Mn掺杂ZnS量子点。
(2)室温磷光检测
在10毫升的比色管中,首先加入1毫升pH 7.2的0.1 mol/L羟乙基哌嗪乙硫磺酸(HEPES)缓冲溶液,然后加入20微升20克/升PEI包覆的锰掺杂氧化锌硫量子点水溶液。将溶液用水稀释至10毫升定容,并充分摇匀。随后逐个加入不同浓度的GTP水溶液,在静置3分钟后使用激发波长为312纳米检测溶液的磷光。
(3)癌细胞提取液的制备
肝癌细胞中鸟苷-5'-三磷酸二钠盐按以下步骤提取:(1)HepG2细胞的复苏。从液氮罐中取出冻存管,迅速放入37 ℃水浴中使其溶解,加入一定量的培养液,800 r/min离心3 min,弃去上清液,加入培养液4 mL,吹打使细胞悬浮。重复上述步骤3次,然后将含细胞的培养液移入培养瓶中。细胞密度控制在约5×104个/mL,放入37 ℃ CO2培养箱中培养;(2)HepG2细胞的培养。培养24 h后更换培养基,如果镜检观察发现细胞数量很多,则进行传代;(3)HepG2细胞提取液的制备。首先选择体外指数生长的细胞,消化后用血球计进行计数,总量为2.5×107个/mL,然后以800 r/min离心3 min,并用预冷的PBS洗3次,800 r/min离心3 min,重悬于0.25 mL预冷的超纯水中,在0 ℃超声降解20 min使细胞壁破裂。为去除溶解产物中的细胞碎片和蛋白,依次用Amicon Ultra-4离心膜30 kDa和3 kDa离心分离(10 000 r/min×15 min)。将得到的癌细胞提取液准确稀释20倍用于检测。
2. 方法二
脱氧核酶具有独特的催化和结构性质,其中一种具有自身磷酸化能力的脱氧核酶DK2,在锰(II)存在下可将GTP上的一个磷酸基转移到5′端,而λ外切酶(λexo)可催化5′端磷酸化的双链DNA分子从5′逐渐水解到3′,但不能裂解5′-OH端。荧光染料SYBR Green I (SG I)能与双链DNA结合并产生强荧光,但与单链DNA混合时只能发出微弱的荧光。Chengzhen Hu提出了一种基于脱氧核酶DK2自身磷酸化和λexo特异性水解的新型GTP非标记荧光检测方法。该方法具有操作简便、灵敏度高、特异性好、成本低、无需荧光团(猝灭基团)标记等优点,在生物应用中具有巨大的潜力。
参考:
[1]任呼博,杨成雄,严秀平.微波-超声波辅助合成聚乙烯亚胺包覆Mn掺杂ZnS量子点用于室温磷光检测三磷酸鸟苷[J].分析测试学报,2012,31(09):1042-1050.
[2]Hu C, Jiang K, Shao Z, et al. A DNAzyme-based label-free fluorescent probe for guanosine-5′-triphosphate detection[J]. Analyst, 2020, 145(21): 6948-6954.
1
本文将介绍如何测定药物中京尼平苷酸的含量,京尼平苷酸是一种常见的药物成分,在药品分析中具有重要的意义。通过详细讨论分析方法和技术步骤,我们可以更好地了解药物质量控制中的关键环节和操作要点。
简述:京尼平苷酸为环烯醚萜类化合物,在车前子、杜仲中含量较高,为杜仲中主要有效活性成分之一。有研究表明京尼平苷酸具有保肝利胆、修复损伤脊髓、提高记忆力、抗炎等多种生物活性。京尼平苷酸为京尼平苷一相代谢产物,两者结构相似,前者有抗炎活性。目前,京尼平苷酸的研究集中在含量测定、提取纯化、生物活性方面。

含量测定:
1. 报道一
王开裕等人建立了高效液相色谱法测定金匮肾气丸中马钱苷、京尼平苷酸、23-乙酰泽泻醇B和β-蜕皮甾酮的含量。方法具体为:将金匮肾气丸样品研细、甲醇超声提取后,用Capcell Pak UG C18色谱柱分离,柱温为28℃,京尼平苷酸、β-蜕皮甾酮、马钱苷和23-乙酰泽泻醇B检测波长分别设定为254、250、240、254 nm,进样体积为10μL,流速为1.0 mL·min-1,以乙腈为流动相A、3 mL·L-1磷酸溶液为流动相B,梯度洗脱,外标法定量检测。
京尼平苷酸、β-蜕皮甾酮、马钱苷和23-乙酰泽泻醇B质量浓度分别在0.049 5~4.95、0.098 1~9.81、0.019 7~1.97、0.098 3~9.83μg·mL-1范围内具有良好的线性关系;重复性RSD值(n=6)分别为1.62%、1.31%、1.76%、1.24%;平均加样回收率分别为100.05%、98.33%、99.97%、99.42%。该方法具有前处理简单、分析时间短、检测结果准确等优点,适用于金匮肾气丸的质量控制。
2. 报道二
闫占宽等人采用超高效液相色谱串联四极杆静电场轨道阱质谱法(UPLC-Q-Exactive Orbitrap-MS/MS)对五神汤中的化学成分进行鉴定,进而对五神汤中京尼平苷酸、秦皮甲素、咖啡酸、木犀草苷、β-蜕皮甾酮和毛蕊花糖苷6种成分进行含量测定。方法具体为:UPLC-Q-Exactive Orbitrap-MS/MS法鉴定五神汤中的化学成分,采用ACQUIFY UPLCTMBEH C18色谱柱(100 mm×2.1 mm,1.7μm)进行分离,以0.1%甲酸-乙腈为流动相进行梯度洗脱,检测波长为254 nm,柱温30℃;电喷雾离子源(ESI)全扫模式;根据一级质谱中的分子离子,推测化合物的相对分子质量和元素组成;再利用二级质谱信息,与对照品及文献报道中化合物信息库进行比对,推测五神汤的体外化学成分。HPLC法检测五神汤中京尼平苷酸、秦皮甲素、咖啡酸、木犀草苷、β-蜕皮甾酮和毛蕊花糖苷,Aglient Eclipse XDB-C18色谱柱(150 mm×4.6 mm,5 mm),流动相为0.1%磷酸水(A)-乙腈(B),梯度洗脱,检测波长254 nm,体积流量1.0 mL·min-1,柱温30℃,进样量10μL。
研究共检测到五神汤中的71种化学成分,主要包括黄酮类、酚酸类、三萜皂苷类以及香豆素类;6种成分在浓度范围内线性关系良好,平均加样回收率为89.6%~93.0%。京尼平苷酸、秦皮甲素、咖啡酸、木犀草苷、β-蜕皮甾酮和毛蕊花糖苷在10批样品中的平均质量浓度依次为2.92、2.48、1.72、1.38、2.08、1.77 mg·g-1。建立的UPLC-Q-Exactive Orbitrap-MS/MS、HPLC法可用于五神汤化学成分的鉴定和含量测定。
3. 报道三
那效旗等人建立了RP-HPLC-UV色谱法测定清心莲子饮的基准物质中京尼平苷酸的含量。方法具体为:采用依利特C18柱(Sinochrom ODS-BP,4.6mm×200mm,5μm),0.5%的HAc溶液和甲醇进行梯度洗脱,流速控制为1.0m L·min-1,在254nm,柱温25℃下进样,进样量为10μL。
京尼平苷酸的含量与色谱峰面积呈现出良好的线性关系,其线性方程为Y=11.185X+0.065(R2=0.9999)。精密度、重复性和加样回收率等试验RSD均小于3%,平均加样回收率为101.05%,RSD为2.45%。该含量测定的方法可行,操作简易,灵敏度高,适于清心莲子饮的物质基准中京尼平苷酸的含量测定,可作为清心莲子饮质量评价的方法。
4. 报道四
闫桦等人建立能同时测定排石颗粒中木通苯乙醇苷B、京尼平苷酸和丹皮酚含量的RP-HPLC法。方法具体为:将排石颗粒研细后,加甲醇超声提取,滤过。以Agilent eclipse zorbax SB-C18色谱柱分离;检测波长:254 nm (京尼平苷酸)、273 nm (丹皮酚)和330nm(木通苯乙醇苷B);流速1.0 mL min-1;柱温30℃;进样量10 μL;流动相A:甲醇,流动相B:0.5%冰乙酸溶液,采用梯度洗脱;二极管阵列检测器定量检测。
京尼平苷酸、木通苯乙醇苷B和丹皮酚依次在质量浓度0.981~98.1、0.393~39.3和0.199~19.9mg·L-1内有良好的线性,重复性RSD为2.61%、1.49%和2.02%,平均加标回收率依次为100.14%、99.70%和100.27%,溶液稳定性和仪器精密度均良好。该方法具有快速、准确等特点,适用于排石颗粒的质量控制。
参考:
[1]王开裕,林天东,吴振宁,等. 金匮肾气丸中马钱苷、京尼平苷酸、23-乙酰泽泻醇B和β-蜕皮甾酮含量测定方法的建立 [J]. 西北药学杂志, 2023, 38 (05): 48-52.
[2]闫占宽,张敏,刘钦伟,等. 五神汤化学成分鉴定及京尼平苷酸、秦皮甲素、咖啡酸、木犀草苷、β-蜕皮甾酮和毛蕊花糖苷定量检测 [J]. 药物评价研究, 2023, 46 (05): 1047-1056.
[3]那效旗,陈忠新,苟鑫宇,等. RP-HPLC-UV法测定清心莲子饮基准物质中京尼平苷酸的含量 [J]. 化学工程师, 2021, 35 (12): 20-22+64. DOI:10.16247/j.cnki.23-1171/tq.20211220.
[4]肖晓燕,冯果,李玮,等. UPLC-MS/MS分析京尼平苷酸在大鼠体内的组织分布研究 [J]. 贵州医药, 2021, 45 (08): 1182-1185.
[5]闫桦,齐赛卿,王晶,等. RP-HPLC法同时测定排石颗粒中木通苯乙醇苷B、京尼平苷酸和丹皮酚的含量 [J]. 中国临床药学杂志, 2021, 30 (04): 285-288. DOI:10.19577/j.1007-4406.2021.04.010.
显示全部本文将介绍如何测定药物中京尼平苷酸的含量,京尼平苷酸是一种常见的药物成分,在药品分析中具有重要的意义。通过详细讨论分析方法和技术步骤,我们可以更好地了解药物质量控制中的关键环节和操作要点。
简述:京尼平苷酸为环烯醚萜类化合物,在车前子、杜仲中含量较高,为杜仲中主要有效活性成分之一。有研究表明京尼平苷酸具有保肝利胆、修复损伤脊髓、提高记忆力、抗炎等多种生物活性。京尼平苷酸为京尼平苷一相代谢产物,两者结构相似,前者有抗炎活性。目前,京尼平苷酸的研究集中在含量测定、提取纯化、生物活性方面。
含量测定:
1. 报道一
王开裕等人建立了高效液相色谱法测定金匮肾气丸中马钱苷、京尼平苷酸、23-乙酰泽泻醇B和β-蜕皮甾酮的含量。方法具体为:将金匮肾气丸样品研细、甲醇超声提取后,用Capcell Pak UG C18色谱柱分离,柱温为28℃,京尼平苷酸、β-蜕皮甾酮、马钱苷和23-乙酰泽泻醇B检测波长分别设定为254、250、240、254 nm,进样体积为10μL,流速为1.0 mL·min-1,以乙腈为流动相A、3 mL·L-1磷酸溶液为流动相B,梯度洗脱,外标法定量检测。
京尼平苷酸、β-蜕皮甾酮、马钱苷和23-乙酰泽泻醇B质量浓度分别在0.049 5~4.95、0.098 1~9.81、0.019 7~1.97、0.098 3~9.83μg·mL-1范围内具有良好的线性关系;重复性RSD值(n=6)分别为1.62%、1.31%、1.76%、1.24%;平均加样回收率分别为100.05%、98.33%、99.97%、99.42%。该方法具有前处理简单、分析时间短、检测结果准确等优点,适用于金匮肾气丸的质量控制。
2. 报道二
闫占宽等人采用超高效液相色谱串联四极杆静电场轨道阱质谱法(UPLC-Q-Exactive Orbitrap-MS/MS)对五神汤中的化学成分进行鉴定,进而对五神汤中京尼平苷酸、秦皮甲素、咖啡酸、木犀草苷、β-蜕皮甾酮和毛蕊花糖苷6种成分进行含量测定。方法具体为:UPLC-Q-Exactive Orbitrap-MS/MS法鉴定五神汤中的化学成分,采用ACQUIFY UPLCTMBEH C18色谱柱(100 mm×2.1 mm,1.7μm)进行分离,以0.1%甲酸-乙腈为流动相进行梯度洗脱,检测波长为254 nm,柱温30℃;电喷雾离子源(ESI)全扫模式;根据一级质谱中的分子离子,推测化合物的相对分子质量和元素组成;再利用二级质谱信息,与对照品及文献报道中化合物信息库进行比对,推测五神汤的体外化学成分。HPLC法检测五神汤中京尼平苷酸、秦皮甲素、咖啡酸、木犀草苷、β-蜕皮甾酮和毛蕊花糖苷,Aglient Eclipse XDB-C18色谱柱(150 mm×4.6 mm,5 mm),流动相为0.1%磷酸水(A)-乙腈(B),梯度洗脱,检测波长254 nm,体积流量1.0 mL·min-1,柱温30℃,进样量10μL。
研究共检测到五神汤中的71种化学成分,主要包括黄酮类、酚酸类、三萜皂苷类以及香豆素类;6种成分在浓度范围内线性关系良好,平均加样回收率为89.6%~93.0%。京尼平苷酸、秦皮甲素、咖啡酸、木犀草苷、β-蜕皮甾酮和毛蕊花糖苷在10批样品中的平均质量浓度依次为2.92、2.48、1.72、1.38、2.08、1.77 mg·g-1。建立的UPLC-Q-Exactive Orbitrap-MS/MS、HPLC法可用于五神汤化学成分的鉴定和含量测定。
3. 报道三
那效旗等人建立了RP-HPLC-UV色谱法测定清心莲子饮的基准物质中京尼平苷酸的含量。方法具体为:采用依利特C18柱(Sinochrom ODS-BP,4.6mm×200mm,5μm),0.5%的HAc溶液和甲醇进行梯度洗脱,流速控制为1.0m L·min-1,在254nm,柱温25℃下进样,进样量为10μL。
京尼平苷酸的含量与色谱峰面积呈现出良好的线性关系,其线性方程为Y=11.185X+0.065(R2=0.9999)。精密度、重复性和加样回收率等试验RSD均小于3%,平均加样回收率为101.05%,RSD为2.45%。该含量测定的方法可行,操作简易,灵敏度高,适于清心莲子饮的物质基准中京尼平苷酸的含量测定,可作为清心莲子饮质量评价的方法。
4. 报道四
闫桦等人建立能同时测定排石颗粒中木通苯乙醇苷B、京尼平苷酸和丹皮酚含量的RP-HPLC法。方法具体为:将排石颗粒研细后,加甲醇超声提取,滤过。以Agilent eclipse zorbax SB-C18色谱柱分离;检测波长:254 nm (京尼平苷酸)、273 nm (丹皮酚)和330nm(木通苯乙醇苷B);流速1.0 mL min-1;柱温30℃;进样量10 μL;流动相A:甲醇,流动相B:0.5%冰乙酸溶液,采用梯度洗脱;二极管阵列检测器定量检测。
京尼平苷酸、木通苯乙醇苷B和丹皮酚依次在质量浓度0.981~98.1、0.393~39.3和0.199~19.9mg·L-1内有良好的线性,重复性RSD为2.61%、1.49%和2.02%,平均加标回收率依次为100.14%、99.70%和100.27%,溶液稳定性和仪器精密度均良好。该方法具有快速、准确等特点,适用于排石颗粒的质量控制。
参考:
[1]王开裕,林天东,吴振宁,等. 金匮肾气丸中马钱苷、京尼平苷酸、23-乙酰泽泻醇B和β-蜕皮甾酮含量测定方法的建立 [J]. 西北药学杂志, 2023, 38 (05): 48-52.
[2]闫占宽,张敏,刘钦伟,等. 五神汤化学成分鉴定及京尼平苷酸、秦皮甲素、咖啡酸、木犀草苷、β-蜕皮甾酮和毛蕊花糖苷定量检测 [J]. 药物评价研究, 2023, 46 (05): 1047-1056.
[3]那效旗,陈忠新,苟鑫宇,等. RP-HPLC-UV法测定清心莲子饮基准物质中京尼平苷酸的含量 [J]. 化学工程师, 2021, 35 (12): 20-22+64. DOI:10.16247/j.cnki.23-1171/tq.20211220.
[4]肖晓燕,冯果,李玮,等. UPLC-MS/MS分析京尼平苷酸在大鼠体内的组织分布研究 [J]. 贵州医药, 2021, 45 (08): 1182-1185.
[5]闫桦,齐赛卿,王晶,等. RP-HPLC法同时测定排石颗粒中木通苯乙醇苷B、京尼平苷酸和丹皮酚的含量 [J]. 中国临床药学杂志, 2021, 30 (04): 285-288. DOI:10.19577/j.1007-4406.2021.04.010.
1
本文将探讨如何测定药物中龙胆苦苷的含量,这是一项关键的药物分析任务,有助于确保药品质量和安全性。通过介绍分析方法和技术步骤,我们可以更准确地确定药物中龙胆苦苷的含量,为药品监管和质量控制提供重要参考。
简述:龙胆草在中药制剂中应用广泛,用量大。现行药典规定龙胆草的药用部分为干根,而大量的茎、叶、花被丢弃。

研究发现,根和根茎中含有裂环醚萜苷、生物碱、类黄酮、类固醇、香豆素和内酯等化合物。其中,龙胆苦苷含量最高,被中国药典定为质量标准控制的目标成分。龙胆苦苷是一种环醚萜苷类化合物,现代药理研究表明其具有抗抑郁、保肝、抗肿瘤、抗炎、糖尿病和骨质疏松作用。
含量测定:
1. 报道一
吴勇等人建立高效液相色谱法同时测定金胆片中4种主要活性成分含量的方法。研究采用Agilent Eclipse X DB C18(4.6 mm×250 mm,5μm)色谱柱;以乙腈(A)-0.1%磷酸溶液(B)为流动相梯度洗脱,检测波长为270 nm;柱温为30℃;流速1.0 mL·min-1。
龙胆苦苷、虎杖苷、槲皮素和大黄素质量浓度分别在7.875~78.75μg·mL-1(r=0.999 9)、6.75~67.50μg·mL-1(r=0.999 7)、7.726~77.26μg·mL-1(r=0.999 4)、3.809~38.09μg·mL-1(r=0.999 8)范围内与峰面积呈良好的线性关系,平均加样回收率分别为99.31%、99.21%、99.04%、99.59%(n=6),RSD分别为1.86%、1.24%、1.37%、1.15%。该实验方法准确性可靠、重复性和稳定性良好,专属性强,可为金胆片的质量控制和标准提升提供参考依据。
2. 报道二
江涛等人建立了同时测定追风透骨丸中龙胆苦苷和芍药苷含量的高效液相色谱法。研究采用的色谱柱为Agilent Eclipse Plus C18柱(250 mm×4.6 mm,5μm),流动相为甲醇-0.1%磷酸溶液(30∶70,V/V),流速为1.0 mL/min,检测波长为230 nm,柱温为30℃,进样量为10μL。
龙胆苦苷和芍药苷质量浓度分别在15.718~157.175μg/mL和16.095~160.949μg/mL范围内与峰面积线性关系良好(r=0.999 9,n=6);精密度、稳定性、重复性试验结果的RSD均小于2.0%;平均加样回收率分别为99.77%和99.28%,RSD分别为0.66%和0.60%(n=9)。12批样品中龙胆苦苷和芍药苷的平均含量分别为0.856,1.174 mg/g。该方法操作简便、结果准确、重复性及专属性良好,可用于同时测定追风透骨丸中龙胆苦苷和芍药苷的含量。
3. 报道三
眭荣春等人建立HPLC法测定蕲蛇药酒中马钱苷酸和龙胆苦苷的含量。研究采用Ultimate Plus C18色谱柱(250 mm×4.6 mm,5μm),流动相为乙腈(A)-0.1%磷酸溶液(B),梯度洗脱,流速为1.0 mL/min,检测波长为254 nm,柱温为30℃,进样量为10μL。
马钱苷酸和龙胆苦苷分别在浓度为0.012 60 mg/mL~0.314 9 mg/mL和0.020 17 mg/mL~0.504 2 mg/mL范围内呈良好的线性关系,平均加样回收率分别为100.19%、96.62%,RSD均小于2.0%。该HPLC法简便、准确,重复性好,可用于测定蕲蛇药酒中马钱苷酸和龙胆苦苷的含量。
4. 报道四
安艳苏等人建立了高效液相色谱法测定和血明目片中龙胆苦苷、木犀草苷、黄芩苷3种成分的含量。研究采用Hypersil ODS2色谱柱(4.6 mm×250 mm,5 μm);柱温:30 ℃;流动相:0.1%磷酸溶液-乙腈,梯度洗脱;流速:1.0 mL/min;检测波长:254 nm。
和血明目片的最佳提取方式为甲醇50 mL超声提取30 min。龙胆苦苷、木犀草苷、黄芩苷的线性范围分别为8.04 μg/mL~80.40 μg/mL(r=0.999 5)、3.28 μg/mL~32.80 μg/mL(r=0.999 8)、10.64 μg/mL~106.37 μg/mL(r=0.999 0);加样回收率的范围在96.50%~102.13%之间;稳定性、重复性试验的RSD值均小于2.3%。该法专属性强、操作简便、结果准确,稳定性好,可用于和血明目片中龙胆苦苷、木犀草苷、黄芩苷的含量测定。
5. 报道五
张丽娟等人建立了同时测定鼻咽清颗粒中龙胆苦苷、迷迭香酸和蒙花苷3种活性成分含量的反相高效液相色谱法。研究采用的色谱柱为ACE Excel C18柱(250 mm×4.6 mm,5μm),流动相为乙腈-0.1%磷酸水溶液(梯度洗脱),流速为1.0 mL/min,检测波长为274 nm(0~20 min时龙胆苦苷)、334 nm(20~40 min时迷迭香酸和蒙花苷),柱温为30℃,进样量为10μL。
龙胆苦苷、迷迭香酸、蒙花苷质量浓度分别在5.29~169.34μg/mL(r=1.000 0,n=6),1.00~32.09μg/mL (r=0.999 7,n=6),2.28~72.88μg/mL(r=1.000 0,n=6)范围内与峰面积线性关系良好,精密度、稳定性、重复性试验的RSD均小于2.0%,平均回收率分别为100.18%,98.63%,99.34%,RSD分别为1.47%,2.09%,2.17%(n=9)。所建立的方法能有效控制其中的活性成分,前处理简单,重复性好,准确度高,可用于同时测定鼻咽清颗粒中龙胆苦苷、迷迭香酸和蒙花苷的含量,完善其质量标准。
参考:
[1]吴勇,刘燕,丁茹,等. HPLC法同时测定金胆片中龙胆苦苷等4种主成分的含量 [J]. 中国药品标准, 2024, 25 (01): 94-98. DOI:10.19778/j.chp.2024.01.016.
[2]江涛,陈丽斯,彭红英,等. 高效液相色谱法同时测定追风透骨丸中龙胆苦苷和芍药苷含量 [J]. 中国药业, 2023, 32 (13): 83-86.
[3]孙楷超,段小群. 龙胆苦苷的药理作用机制的研究进展 [J]. 广东化工, 2023, 50 (08): 105-107.
[4]眭荣春,李金林,周玉春,等. HPLC法测定蕲蛇药酒中马钱苷酸和龙胆苦苷的含量 [J]. 药品评价, 2022, 19 (23): 1423-1426. DOI:10.19939/j.cnki.1672-2809.2022.23.04.
[5]安艳苏,毕秋左,张顺平. 高效液相色谱法测定和血明目片中龙胆苦苷、木犀草苷和黄芩苷含量 [J]. 药品评价, 2022, 19 (03): 137-140. DOI:10.19939/j.cnki.1672-2809.2022.03.03.
[6]张丽娟,蓝献泉,郑锦坤,等. 反相高效液相色谱法同时测定鼻咽清颗粒中龙胆苦苷、迷迭香酸和蒙花苷含量 [J]. 中国药业, 2021, 30 (21): 62-65.
[7]https://en.wikipedia.org/wiki/Gentiana_macrophylla
显示全部本文将探讨如何测定药物中龙胆苦苷的含量,这是一项关键的药物分析任务,有助于确保药品质量和安全性。通过介绍分析方法和技术步骤,我们可以更准确地确定药物中龙胆苦苷的含量,为药品监管和质量控制提供重要参考。
简述:龙胆草在中药制剂中应用广泛,用量大。现行药典规定龙胆草的药用部分为干根,而大量的茎、叶、花被丢弃。
研究发现,根和根茎中含有裂环醚萜苷、生物碱、类黄酮、类固醇、香豆素和内酯等化合物。其中,龙胆苦苷含量最高,被中国药典定为质量标准控制的目标成分。龙胆苦苷是一种环醚萜苷类化合物,现代药理研究表明其具有抗抑郁、保肝、抗肿瘤、抗炎、糖尿病和骨质疏松作用。
含量测定:
1. 报道一
吴勇等人建立高效液相色谱法同时测定金胆片中4种主要活性成分含量的方法。研究采用Agilent Eclipse X DB C18(4.6 mm×250 mm,5μm)色谱柱;以乙腈(A)-0.1%磷酸溶液(B)为流动相梯度洗脱,检测波长为270 nm;柱温为30℃;流速1.0 mL·min-1。
龙胆苦苷、虎杖苷、槲皮素和大黄素质量浓度分别在7.875~78.75μg·mL-1(r=0.999 9)、6.75~67.50μg·mL-1(r=0.999 7)、7.726~77.26μg·mL-1(r=0.999 4)、3.809~38.09μg·mL-1(r=0.999 8)范围内与峰面积呈良好的线性关系,平均加样回收率分别为99.31%、99.21%、99.04%、99.59%(n=6),RSD分别为1.86%、1.24%、1.37%、1.15%。该实验方法准确性可靠、重复性和稳定性良好,专属性强,可为金胆片的质量控制和标准提升提供参考依据。
2. 报道二
江涛等人建立了同时测定追风透骨丸中龙胆苦苷和芍药苷含量的高效液相色谱法。研究采用的色谱柱为Agilent Eclipse Plus C18柱(250 mm×4.6 mm,5μm),流动相为甲醇-0.1%磷酸溶液(30∶70,V/V),流速为1.0 mL/min,检测波长为230 nm,柱温为30℃,进样量为10μL。
龙胆苦苷和芍药苷质量浓度分别在15.718~157.175μg/mL和16.095~160.949μg/mL范围内与峰面积线性关系良好(r=0.999 9,n=6);精密度、稳定性、重复性试验结果的RSD均小于2.0%;平均加样回收率分别为99.77%和99.28%,RSD分别为0.66%和0.60%(n=9)。12批样品中龙胆苦苷和芍药苷的平均含量分别为0.856,1.174 mg/g。该方法操作简便、结果准确、重复性及专属性良好,可用于同时测定追风透骨丸中龙胆苦苷和芍药苷的含量。
3. 报道三
眭荣春等人建立HPLC法测定蕲蛇药酒中马钱苷酸和龙胆苦苷的含量。研究采用Ultimate Plus C18色谱柱(250 mm×4.6 mm,5μm),流动相为乙腈(A)-0.1%磷酸溶液(B),梯度洗脱,流速为1.0 mL/min,检测波长为254 nm,柱温为30℃,进样量为10μL。
马钱苷酸和龙胆苦苷分别在浓度为0.012 60 mg/mL~0.314 9 mg/mL和0.020 17 mg/mL~0.504 2 mg/mL范围内呈良好的线性关系,平均加样回收率分别为100.19%、96.62%,RSD均小于2.0%。该HPLC法简便、准确,重复性好,可用于测定蕲蛇药酒中马钱苷酸和龙胆苦苷的含量。
4. 报道四
安艳苏等人建立了高效液相色谱法测定和血明目片中龙胆苦苷、木犀草苷、黄芩苷3种成分的含量。研究采用Hypersil ODS2色谱柱(4.6 mm×250 mm,5 μm);柱温:30 ℃;流动相:0.1%磷酸溶液-乙腈,梯度洗脱;流速:1.0 mL/min;检测波长:254 nm。
和血明目片的最佳提取方式为甲醇50 mL超声提取30 min。龙胆苦苷、木犀草苷、黄芩苷的线性范围分别为8.04 μg/mL~80.40 μg/mL(r=0.999 5)、3.28 μg/mL~32.80 μg/mL(r=0.999 8)、10.64 μg/mL~106.37 μg/mL(r=0.999 0);加样回收率的范围在96.50%~102.13%之间;稳定性、重复性试验的RSD值均小于2.3%。该法专属性强、操作简便、结果准确,稳定性好,可用于和血明目片中龙胆苦苷、木犀草苷、黄芩苷的含量测定。
5. 报道五
张丽娟等人建立了同时测定鼻咽清颗粒中龙胆苦苷、迷迭香酸和蒙花苷3种活性成分含量的反相高效液相色谱法。研究采用的色谱柱为ACE Excel C18柱(250 mm×4.6 mm,5μm),流动相为乙腈-0.1%磷酸水溶液(梯度洗脱),流速为1.0 mL/min,检测波长为274 nm(0~20 min时龙胆苦苷)、334 nm(20~40 min时迷迭香酸和蒙花苷),柱温为30℃,进样量为10μL。
龙胆苦苷、迷迭香酸、蒙花苷质量浓度分别在5.29~169.34μg/mL(r=1.000 0,n=6),1.00~32.09μg/mL (r=0.999 7,n=6),2.28~72.88μg/mL(r=1.000 0,n=6)范围内与峰面积线性关系良好,精密度、稳定性、重复性试验的RSD均小于2.0%,平均回收率分别为100.18%,98.63%,99.34%,RSD分别为1.47%,2.09%,2.17%(n=9)。所建立的方法能有效控制其中的活性成分,前处理简单,重复性好,准确度高,可用于同时测定鼻咽清颗粒中龙胆苦苷、迷迭香酸和蒙花苷的含量,完善其质量标准。
参考:
[1]吴勇,刘燕,丁茹,等. HPLC法同时测定金胆片中龙胆苦苷等4种主成分的含量 [J]. 中国药品标准, 2024, 25 (01): 94-98. DOI:10.19778/j.chp.2024.01.016.
[2]江涛,陈丽斯,彭红英,等. 高效液相色谱法同时测定追风透骨丸中龙胆苦苷和芍药苷含量 [J]. 中国药业, 2023, 32 (13): 83-86.
[3]孙楷超,段小群. 龙胆苦苷的药理作用机制的研究进展 [J]. 广东化工, 2023, 50 (08): 105-107.
[4]眭荣春,李金林,周玉春,等. HPLC法测定蕲蛇药酒中马钱苷酸和龙胆苦苷的含量 [J]. 药品评价, 2022, 19 (23): 1423-1426. DOI:10.19939/j.cnki.1672-2809.2022.23.04.
[5]安艳苏,毕秋左,张顺平. 高效液相色谱法测定和血明目片中龙胆苦苷、木犀草苷和黄芩苷含量 [J]. 药品评价, 2022, 19 (03): 137-140. DOI:10.19939/j.cnki.1672-2809.2022.03.03.
[6]张丽娟,蓝献泉,郑锦坤,等. 反相高效液相色谱法同时测定鼻咽清颗粒中龙胆苦苷、迷迭香酸和蒙花苷含量 [J]. 中国药业, 2021, 30 (21): 62-65.
[7]https://en.wikipedia.org/wiki/Gentiana_macrophylla
5
考虑到泄漏危险和停机腐蚀,最好衬氟磁力泵。
显示全部
1
引言:
荧光增白剂71是一种广泛应用于纺织品、纸张和塑料中的化学添加剂,旨在提升材料的亮度和白度。为了确保其使用的效果和安全性,准确检测荧光增白剂71的含量和分布至关重要。本文将介绍其检测方法,帮助读者了解如何通过科学手段对荧光增白剂71进行有效检测,从而确保产品质量和安全性。
背景:
荧光增白剂是一种荧光染料,或称为白色染料,也是一种应用广泛的化工原料。荧光增白剂的增白是光学上的补色、增亮,可以显著提高白色基质的白度、浅色基质的鲜艳度,而不会对被增白的基质造成损伤。该法比传统的加蓝增白法和化学漂白法简单,是目前最有效和最常用的提高基质白度的方法之一,在造纸、洗涤剂、化妆品、塑料制品行业中均有着广泛应用。目前全世界生产的荧光增白剂已超过1 000 多种,年总产量达15 万t以上,其分子结构类型有15 种以上。有研究表明,荧光增白剂的毒性与其分子结构有很大关系,目前常用荧光增白剂的毒理数据表明:在适量的添加范围内,荧光增白剂低毒或无毒,但当荧光增白剂含量过高或添加了具有毒性的荧光增白剂时,荧光增白剂就会通过食物的迁移进入人体或直接与人体接触而对人体健康构成潜在威胁。很多国内外标准及法律法规对荧光增白剂在食品接触用纸中的使用有一定的限制要求,荧光增白剂的检测方法也是当前研究的热点之一。
荧光增白剂71,英文名称:Fluorescent Brightener 71,CAS:16090-02-1,分子式:C40H38N12Na2O8S2。荧光增白剂71用作棉纤维、肥皂和洗涤剂的染料;主要用于(大于90%)家用洗涤剂;也用于造纸和纺织工业。
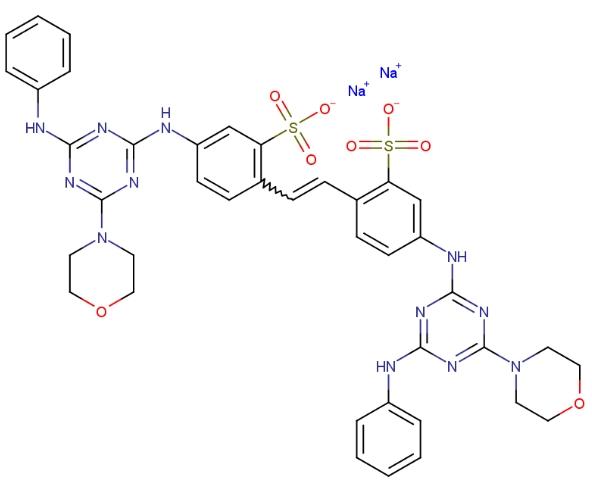
检测研究:
1. 洗衣液、洗衣粉等洗涤用品中荧光增白剂71
冼燕萍等人建立了固相萃取净化结合超高效液相色谱-二极管阵列检测器(UPLC-DAD)同时检测洗衣液、洗衣粉等洗涤用品中荧光增白剂351、85、28和71的分析方法。样品经2%(体积分数)甲酸水溶液-甲醇提取,经WAX固相萃取小柱净化后,采用Phenomenex Synergi Max-RP色谱柱(150 mm×2.0 mm),以乙腈-10 mmol/L乙酸铵为流动相实现待测物(包括顺式和反式异构体)的良好分离,以二极管阵列检测器检测,结合保留时间和光谱图定性,以标准曲线定量。
结果表明,4种荧光增白剂在0.05 ~ 180 mg/L范围内线性关系良好,相关系数均大于0.999 3;方法定量限(S/N=10)为1.5~15 mg/kg;添加水平为5~1 500 mg/kg时,回收率为84.9% ~ 105%,相对标准偏差(RSD,n=6)为3.2% ~6.1%.应用本方法分析了15个样品,阳性样品检出率为53.3%。该法前处理简单,回收率高,精密度好,适用于洗涤用品中4种荧光增白剂的测定。
2. 湿巾中荧光增白剂71
魏峰等人建立了高效液相色谱-二极管阵列检测法分离测定湿巾中三种荧光增白剂FWA 220、FWA 113和FWA 71等物质的方法。采用Eclipse XDB-C18(4.6 mm ×250 mm,5μm)色谱柱,流动相为乙腈-5 mol/L醋酸铵溶液进行梯度洗脱,检测波长为350 nm。实验结果表明,三种荧光增白剂在0.01~400.00 mg/L范围内具有良好的线性关系(相关系数均大于0.9995),检出限和定量限分别在0.01~0.10 mg/L和0.5~5.0 mg/kg范围内。添加水平在10~800 mg/kg时,回收率范围为91.4%~102.2%,相对标准偏差(n=6)为1.6%~5.1%。该方法精密度高,重现性好,结果准确可靠,为荧光增白剂FWA 220、FWA 113和FWA 71的质量评价提供了有效方法。
3. 化妆品中荧光增白剂71
吴震等人建立了通过荧光快筛、高效液相色谱检测和质谱联用确证测定化妆品中4种荧光增白剂的方法。样品经荧光快筛,疑似品经乙腈溶液超声提取,以梯度洗脱和C18色谱柱分离,DAD检测器检测计算荧光增白剂28号、71号、85号和351号含量,阳性品用HPLC - MS/MS确证。在5 ~ 100 μg/mL与1 ~ 20 μg/mL范围内样品中荧光增白剂质量浓度与350 nm紫外光吸收强度呈良好线性关系,相关系数大于0.999,方法对荧光增白剂28号、71号、85号的检出和定量下限分别为10 ng和25 ng,对荧光增白剂 351号的检出和定量下限分别为2 ng和5 ng,回收率91.7% ~ 124.5%,相对标准偏差为0.7% ~ 10.0%。该方法适于快速、高效、准确测定大批化妆品中4种荧光增白剂含量。
4. 食用菌中荧光增白剂71
张建莹等人建立新鲜食用菌中荧光增白剂85(fluorescent brightener VBL, VBL)、荧光增白剂71(fluorescent brightener CXT, CXT)和荧光增白剂113(blankophor BA, BA)的超高效液相色谱-串联质谱(ultra performance liquid chromatography-tandem mass spectrometry, UPLC-MS/MS,)定量检测方法。方法采用60%甲醇-水溶液,75℃水浴振荡提取试样中的荧光增白剂,经弱阴离子交换固相萃取柱净化,采用UPLC-MS/MS在负离子模式下检测,内标法定量。
该方法在0.0100~0.500 mg/L范围内有良好的线性关系,相关系数r2>0.990,在空白新鲜食用菌基质中,不同添加水平下方法的平均回收率范围为80.0%~101%,相对标准偏差为3.4%~11.9%;测定低限为0.100 mg/kg。该方法灵敏度高、准确、重现性好,适用于新鲜食用菌中荧光增白剂85、荧光增白剂71和荧光增白剂113的检测。
5. 食品包装材料中荧光增白剂71
王天娇等人选取浙江省内采集的纸质食品包装材料作为试样,将试样充分粉碎后多次提取、离心,提取液经超高效液相色谱(UPLC)上机分析。采用乙腈/甲醇(2∶3,V/V)混合溶液和含25mmol四丁基溴化铵(TBA)的甲醇水溶液(5∶95,V/V)作为流动相进行梯度洗脱分离,使用ACQUITY UPLC BEH C18色谱柱,建立同时测定11种荧光增白剂(包括荧光增白剂71等)的超高效液相色谱串联荧光检测器和二极管阵列检测器(UPLC-FLD/PDA)双检测器联用的定性定量方法。
结果表明,11种荧光增白剂均得到了良好的分离,荧光检测器(FLD)和二极管阵列检测器(PDA)在25~1000ng/mL线性范围内,其相关系数均大于0.99,加标回收率81.1%~105.7%,RSD均小于10%(n=6)。按仪器的3倍信噪比计算检出限,10倍的信噪比计算定量限,11种荧光增白剂的检出限:荧光检测器2.0~2.8ng/mL,二极管阵列检测器14~25ng/mL。11种荧光增白剂的定量限:荧光检测器6.7~9.3ng/mL,二极管阵列检测器47~83ng/mL。说明该方法能满足实际工作的需要,是日常测定纸质食品包装材料中荧光增白剂的高效定量方法。
参考:
[1] 冼燕萍,郭新东,罗海英,等. 固相萃取-超高效液相色谱分离测定洗涤用品中4种荧光增白剂[J]. 色谱,2013,31(2):162-169. DOI:10.3724/SP.J.1123.2012.09041.
[2] 魏峰,吴际萍,殷祥刚,等. 高效液相色谱法分离测定湿巾中的荧光增白剂[J]. 产业用纺织品,2015(6):41-44. DOI:10.3969/j.issn.1004-7093.2015.06.011.
[3] 吴震,李扬杰,孙清萍. 高效液相色谱法检测化妆品中4种荧光增白剂及其荧光快筛和质谱确证[J]. 香料香精化妆品,2017(6):51-56. DOI:10.3969/j.issn.1000-4475.2017.06.012.
[4] 张建莹,肖锋,叶刚,等. 超高效液相色谱-串联质谱法检测食用菌中荧光增白剂[J]. 食品安全质量检测学报,2014(9):2682-2688.
[5] 王天娇. 食品包装材料中多种荧光增白剂的高效检测技术研究[D]. 浙江:浙江大学,2017.
[6]王海涛,张小慧,曲志勇,等.荧光增白剂检测方法研究进展[J].化学分析计量,2016,25(02):100-102.
显示全部引言:
荧光增白剂71是一种广泛应用于纺织品、纸张和塑料中的化学添加剂,旨在提升材料的亮度和白度。为了确保其使用的效果和安全性,准确检测荧光增白剂71的含量和分布至关重要。本文将介绍其检测方法,帮助读者了解如何通过科学手段对荧光增白剂71进行有效检测,从而确保产品质量和安全性。
背景:
荧光增白剂是一种荧光染料,或称为白色染料,也是一种应用广泛的化工原料。荧光增白剂的增白是光学上的补色、增亮,可以显著提高白色基质的白度、浅色基质的鲜艳度,而不会对被增白的基质造成损伤。该法比传统的加蓝增白法和化学漂白法简单,是目前最有效和最常用的提高基质白度的方法之一,在造纸、洗涤剂、化妆品、塑料制品行业中均有着广泛应用。目前全世界生产的荧光增白剂已超过1 000 多种,年总产量达15 万t以上,其分子结构类型有15 种以上。有研究表明,荧光增白剂的毒性与其分子结构有很大关系,目前常用荧光增白剂的毒理数据表明:在适量的添加范围内,荧光增白剂低毒或无毒,但当荧光增白剂含量过高或添加了具有毒性的荧光增白剂时,荧光增白剂就会通过食物的迁移进入人体或直接与人体接触而对人体健康构成潜在威胁。很多国内外标准及法律法规对荧光增白剂在食品接触用纸中的使用有一定的限制要求,荧光增白剂的检测方法也是当前研究的热点之一。
荧光增白剂71,英文名称:Fluorescent Brightener 71,CAS:16090-02-1,分子式:C40H38N12Na2O8S2。荧光增白剂71用作棉纤维、肥皂和洗涤剂的染料;主要用于(大于90%)家用洗涤剂;也用于造纸和纺织工业。
检测研究:
1. 洗衣液、洗衣粉等洗涤用品中荧光增白剂71
冼燕萍等人建立了固相萃取净化结合超高效液相色谱-二极管阵列检测器(UPLC-DAD)同时检测洗衣液、洗衣粉等洗涤用品中荧光增白剂351、85、28和71的分析方法。样品经2%(体积分数)甲酸水溶液-甲醇提取,经WAX固相萃取小柱净化后,采用Phenomenex Synergi Max-RP色谱柱(150 mm×2.0 mm),以乙腈-10 mmol/L乙酸铵为流动相实现待测物(包括顺式和反式异构体)的良好分离,以二极管阵列检测器检测,结合保留时间和光谱图定性,以标准曲线定量。
结果表明,4种荧光增白剂在0.05 ~ 180 mg/L范围内线性关系良好,相关系数均大于0.999 3;方法定量限(S/N=10)为1.5~15 mg/kg;添加水平为5~1 500 mg/kg时,回收率为84.9% ~ 105%,相对标准偏差(RSD,n=6)为3.2% ~6.1%.应用本方法分析了15个样品,阳性样品检出率为53.3%。该法前处理简单,回收率高,精密度好,适用于洗涤用品中4种荧光增白剂的测定。
2. 湿巾中荧光增白剂71
魏峰等人建立了高效液相色谱-二极管阵列检测法分离测定湿巾中三种荧光增白剂FWA 220、FWA 113和FWA 71等物质的方法。采用Eclipse XDB-C18(4.6 mm ×250 mm,5μm)色谱柱,流动相为乙腈-5 mol/L醋酸铵溶液进行梯度洗脱,检测波长为350 nm。实验结果表明,三种荧光增白剂在0.01~400.00 mg/L范围内具有良好的线性关系(相关系数均大于0.9995),检出限和定量限分别在0.01~0.10 mg/L和0.5~5.0 mg/kg范围内。添加水平在10~800 mg/kg时,回收率范围为91.4%~102.2%,相对标准偏差(n=6)为1.6%~5.1%。该方法精密度高,重现性好,结果准确可靠,为荧光增白剂FWA 220、FWA 113和FWA 71的质量评价提供了有效方法。
3. 化妆品中荧光增白剂71
吴震等人建立了通过荧光快筛、高效液相色谱检测和质谱联用确证测定化妆品中4种荧光增白剂的方法。样品经荧光快筛,疑似品经乙腈溶液超声提取,以梯度洗脱和C18色谱柱分离,DAD检测器检测计算荧光增白剂28号、71号、85号和351号含量,阳性品用HPLC - MS/MS确证。在5 ~ 100 μg/mL与1 ~ 20 μg/mL范围内样品中荧光增白剂质量浓度与350 nm紫外光吸收强度呈良好线性关系,相关系数大于0.999,方法对荧光增白剂28号、71号、85号的检出和定量下限分别为10 ng和25 ng,对荧光增白剂 351号的检出和定量下限分别为2 ng和5 ng,回收率91.7% ~ 124.5%,相对标准偏差为0.7% ~ 10.0%。该方法适于快速、高效、准确测定大批化妆品中4种荧光增白剂含量。
4. 食用菌中荧光增白剂71
张建莹等人建立新鲜食用菌中荧光增白剂85(fluorescent brightener VBL, VBL)、荧光增白剂71(fluorescent brightener CXT, CXT)和荧光增白剂113(blankophor BA, BA)的超高效液相色谱-串联质谱(ultra performance liquid chromatography-tandem mass spectrometry, UPLC-MS/MS,)定量检测方法。方法采用60%甲醇-水溶液,75℃水浴振荡提取试样中的荧光增白剂,经弱阴离子交换固相萃取柱净化,采用UPLC-MS/MS在负离子模式下检测,内标法定量。
该方法在0.0100~0.500 mg/L范围内有良好的线性关系,相关系数r2>0.990,在空白新鲜食用菌基质中,不同添加水平下方法的平均回收率范围为80.0%~101%,相对标准偏差为3.4%~11.9%;测定低限为0.100 mg/kg。该方法灵敏度高、准确、重现性好,适用于新鲜食用菌中荧光增白剂85、荧光增白剂71和荧光增白剂113的检测。
5. 食品包装材料中荧光增白剂71
王天娇等人选取浙江省内采集的纸质食品包装材料作为试样,将试样充分粉碎后多次提取、离心,提取液经超高效液相色谱(UPLC)上机分析。采用乙腈/甲醇(2∶3,V/V)混合溶液和含25mmol四丁基溴化铵(TBA)的甲醇水溶液(5∶95,V/V)作为流动相进行梯度洗脱分离,使用ACQUITY UPLC BEH C18色谱柱,建立同时测定11种荧光增白剂(包括荧光增白剂71等)的超高效液相色谱串联荧光检测器和二极管阵列检测器(UPLC-FLD/PDA)双检测器联用的定性定量方法。
结果表明,11种荧光增白剂均得到了良好的分离,荧光检测器(FLD)和二极管阵列检测器(PDA)在25~1000ng/mL线性范围内,其相关系数均大于0.99,加标回收率81.1%~105.7%,RSD均小于10%(n=6)。按仪器的3倍信噪比计算检出限,10倍的信噪比计算定量限,11种荧光增白剂的检出限:荧光检测器2.0~2.8ng/mL,二极管阵列检测器14~25ng/mL。11种荧光增白剂的定量限:荧光检测器6.7~9.3ng/mL,二极管阵列检测器47~83ng/mL。说明该方法能满足实际工作的需要,是日常测定纸质食品包装材料中荧光增白剂的高效定量方法。
参考:
[1] 冼燕萍,郭新东,罗海英,等. 固相萃取-超高效液相色谱分离测定洗涤用品中4种荧光增白剂[J]. 色谱,2013,31(2):162-169. DOI:10.3724/SP.J.1123.2012.09041.
[2] 魏峰,吴际萍,殷祥刚,等. 高效液相色谱法分离测定湿巾中的荧光增白剂[J]. 产业用纺织品,2015(6):41-44. DOI:10.3969/j.issn.1004-7093.2015.06.011.
[3] 吴震,李扬杰,孙清萍. 高效液相色谱法检测化妆品中4种荧光增白剂及其荧光快筛和质谱确证[J]. 香料香精化妆品,2017(6):51-56. DOI:10.3969/j.issn.1000-4475.2017.06.012.
[4] 张建莹,肖锋,叶刚,等. 超高效液相色谱-串联质谱法检测食用菌中荧光增白剂[J]. 食品安全质量检测学报,2014(9):2682-2688.
[5] 王天娇. 食品包装材料中多种荧光增白剂的高效检测技术研究[D]. 浙江:浙江大学,2017.
[6]王海涛,张小慧,曲志勇,等.荧光增白剂检测方法研究进展[J].化学分析计量,2016,25(02):100-102.

|

|
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手




















