https://www.zhihu.com/question/538394912/answer/3353833849 (链接有资料及视频演示,链接官方账号文章栏目内资料有联系方式)
自荐一款优秀产品 新一代接触器零能耗兼具抗晃电解决方案
从芯定义 抗晃电技术,让晃电不在发生。
第五代零能耗型接触器从新定义交流接触器,并延伸实现抗晃电功能,相较于NC2 、CJX2F 、CJX2S、 CJ20、 CJ40、SDDR-DZ7等节电率100%,节电能力800-1501kwh/年;新一代全系列接触器实现零能耗运行,适用于对供电质量要求连续型行业如:工业园区,石油开采、冶金化工、矿业开采、半导体等业态。
适用工频回路和变频器回路,二次设计与常规接触器接线相同,简单便捷,相较于来电自启和储能延时型抗晃电,新一代接触器应用凸轮技术解决前4代产品缺陷(传统电动机保护器应用电压检测技术,控制交流接触器再启动式抗晃电功能和采用直流低压大电流延时交流接触器线圈断开型抗晃电模块、存在集中启动冲击电网隐患、人为紧急停电或配电回路故障存在延时断电,紧急情况下无法瞬时停电保障人身和设备安全等问题),第五代零能耗型抗晃电接触器具备故障自识别能力 仅晃电故障时智能延时保护,人为干预或故障停机接触器瞬时断开,提高应用场景安全性同时具备节能效应,能效等级1+级,年节电800-1501kwh;可实现多路拖动功能,可应用于单一配电回路和箱变多路集成场合 ;
全新一代产品实现晃电保护、安全应用的同时从使用端降低能耗,为您的场景解决高能耗及晃电隐患。
https://www.zhihu.com/question/538394912/answer/3353833849 (链接有资料及视频演示,链接官方账号文章栏目内资料有联系方式)
自荐一款优秀产品 新一代接触器零能耗兼具抗晃电解决方案
从芯定义 抗晃电技术,让晃电不在发生。
第五代零能耗型接触器从新定义交流接触器,并延伸实现抗晃电功能,相较于NC2 、CJX2F 、CJX2S、 CJ20、 CJ40、SDDR-DZ7等节电率100%,节电能力800-1501kwh/年;新一代全系列接触器实现零能耗运行,适用于对供电质量要求连续型行业如:工业园区,石油开采、冶金化工、矿业开采、半导体等业态。
适用工频回路和变频器回路,二次设计与常规接触器接线相同,简单便捷,相较于来电自启和储能延时型抗晃电,新一代接触器应用凸轮技术解决前4代产品缺陷(传统电动机保护器应用电压检测技术,控制交流接触器再启动式抗晃电功能和采用直流低压大电流延时交流接触器线圈断开型抗晃电模块、存在集中启动冲击电网隐患、人为紧急停电或配电回路故障存在延时断电,紧急情况下无法瞬时停电保障人身和设备安全等问题),第五代零能耗型抗晃电接触器具备故障自识别能力 仅晃电故障时智能延时保护,人为干预或故障停机接触器瞬时断开,提高应用场景安全性同时具备节能效应,能效等级1+级,年节电800-1501kwh;可实现多路拖动功能,可应用于单一配电回路和箱变多路集成场合 ;
全新一代产品实现晃电保护、安全应用的同时从使用端降低能耗,为您的场景解决高能耗及晃电隐患。
 1
1
2,3-二溴丁二酸是一种重要的有机合成中间体,其分析和应用在化工领域和合成化学等领域具有重要意义。
简述:2,3-二溴丁二酸,英文名称:meso-2,3-Dibromosuccinic acid,CAS:608-36-6,分子式:C4H4Br2O4,密度:2.486 g/cm3。二溴丁二酸是一种具有极高价值的精细化工产品和成药物中间体,在化工、医药等 行业具有广泛的应用,
1. 分析方法:
2,3二溴丁二酸是生物素合成生产时的一个重要中间体,知道其含量对生产各环节的质量监控和顺利生产具有重要作用。周亚新等人提出一种快速测定2,3二 溴丁二酸的分析方法,即先用0.1mol/L KBrO3-KBr 产生的溴加成后,用碘量法测出富马酸的含量,再用0.1mol/L NaOH测出总酸消耗NaOH的毫升数, 由测出的富马酸含量可得其消耗NaOH的毫升数,由总酸消耗NaOH的毫升数减去富马酸消耗NaOH 的毫升数,即可算出2,3二溴丁二酸含量。实验步骤如下:
(1)富马酸的测定
称取0.5000g的2,3二溴丁二酸中间体样品于250mL碘量瓶中,用少许乙醇溶解,加10mL蒸馏水,再加入0.1mol/L KBrO3-KBr溶液和 5mL浓HCl,密封摇匀,以少许15%KI液封口,放置 5min后,加15mL 15%KI溶液,以水封口,再放置 5min,然后加入30mL蒸馏水,以0.1mol/L Na2S2O3溶液滴定近终点时,加入1mL淀粉指示剂,滴至蓝色消失,同时作一空白试验。
计算:含量%=(V空-V 样)x Mx0.116/(Wx2);V空:空白样品消耗Na 2S2O3的毫升数;V样:样品消耗Na 2S 2O 3的毫升数;W:样品重量(克); 0.116:富马酸毫克分子量;M:硫代硫酸钠的摩尔浓度,mol/L。
(2)总酸测定
称取0.2000g的2,3二溴丁二酸 间体样品于250mL碘量瓶中,用少许乙醇溶解,加 30mL蒸馏水,2-3滴酚酞指示剂,用0.1mol/L NaOH 滴至粉红色,记下总消耗氢氧化钠的毫升数。
该方法简便,易于操作,定量准确,可用于生物素合成过程中间体含量的监控。
2. 应用:
2.1 合成柔性羧酸构筑的具有孔洞结构的配合物
马科芳等人通过对反应体系酸碱度的微调节,合成得到了{[Cu(C4H 2Br2O4)] 1/2[Cu(C4H2Br2O4)(H2O)2]1/2(C6H12N4)(H2O)3}n(1);{[Ni(C4H2Br2O4)] 1/2[Ni(C4H2Br2O4)(H2O)2]1/2(C6H12N4)(H2O)3} n(2); [Ni(C 4H 2Br 2O 4)(C3H3N2)2(H 2O) 2] n(3); [Co(C4H2Br2O4)(C3H3N2)2(H2O)2] n(4); [Zn(C4H2Br2O4)(C3H 3N2)2] n(5); [Cd2(C4H2Br2O4)2(C3H3N2)4(H2O)4] n(6)等六个以2, 3-二溴丁二酸为主要骨架构筑的孔洞配合物。举例配合物({Cu2(C4H2Br 2O4)((C6H12N4)(H2O)4}n(1)的合成
取配体2, 3-二溴丁二酸(0.5mmol, 0.136g)溶于15ml甲醇和水的混合溶液中(2: 1),在此溶液中加入Cu(ClO4)2(0.5mmol, 0.124g),搅拌半小时得到一绿色溶液,在此基础上,逐步滴入5ml的六次甲基四胺(1mmol, 0.140g)水溶液,再搅拌半小时,最后用5%的NaOH调节pH到6.0,过滤静置,两天后得到淡绿色晶体。(产率: 80%)。
2. 合成2,3-二溴丁二酸配位聚合物
翁婉丹等人以meso-2,3-二溴丁二酸(H2DBrS)为原料所形成的10例化合物。下表中列出其重要晶体学参数。
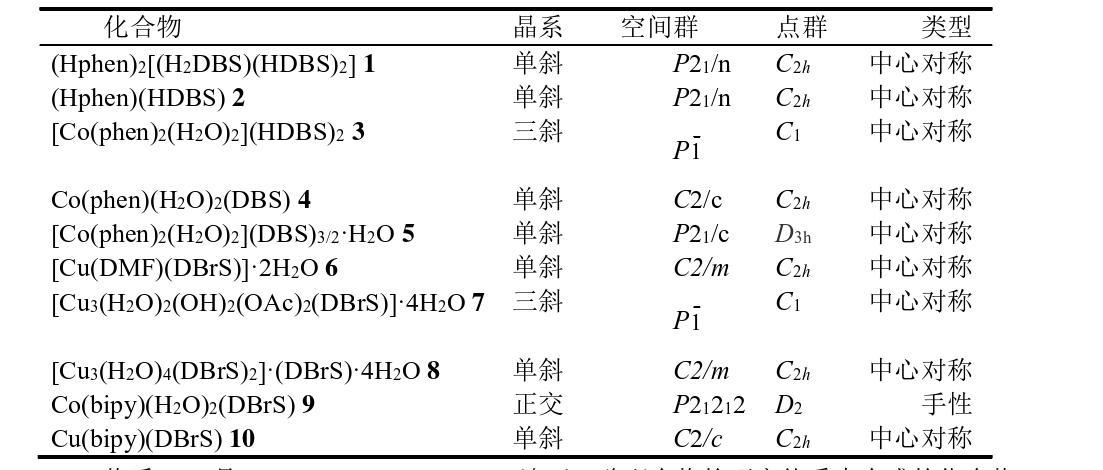
物质1-5是Co(II)-Phenanthroline-二溴丁二酸配合物的研究体系中合成的化合物,配合物6-8是二溴丁二酸H2DBr S与Cu(II)构筑的配合物,9、10是Co(II)/Cu(II)与二溴丁二酸、biby构筑的配合物。列举(Hphen)2[(H 2DBr S)(HDBrS)2] 1 的合成:
称取H2DBrS 0.407 g (1.5 mmol) 溶于10 mL MeOH和10 mL H2O的混合溶剂中,在搅拌下缓慢加phen 0.196 g (1.0 mmol)至上述溶液中,产生白色浑浊。过滤浑浊液,分离得到白色沉淀和无色滤液(pH = 2.04),滤液室温下静置培养,数天后发现有无色片状晶体析出。另称取经干燥后的白色沉淀0.1 g 溶于15 mL MeOH 中进行重结晶,数天后发现有无色片状晶体析出。产率83%(以H2DBrS计)。
参考文献:
[1]翁婉丹. 新型2,3-二溴丁二酸配位聚合物的合成、结构及性质[D]. 宁波大学, 2015.
[2]周亚新. 一种快速测定生物素合成中间体2,3二溴丁二酸的方法 [J]. 福建分析测试, 2010, 19 (03): 36-38.
[3]马科芳. 柔性羧酸构筑的具有孔洞结构的配合物的合成、表征及其性质研究[D]. 温州大学, 2010.
显示全部2,3-二溴丁二酸是一种重要的有机合成中间体,其分析和应用在化工领域和合成化学等领域具有重要意义。
简述:2,3-二溴丁二酸,英文名称:meso-2,3-Dibromosuccinic acid,CAS:608-36-6,分子式:C4H4Br2O4,密度:2.486 g/cm3。二溴丁二酸是一种具有极高价值的精细化工产品和成药物中间体,在化工、医药等 行业具有广泛的应用,
1. 分析方法:
2,3二溴丁二酸是生物素合成生产时的一个重要中间体,知道其含量对生产各环节的质量监控和顺利生产具有重要作用。周亚新等人提出一种快速测定2,3二 溴丁二酸的分析方法,即先用0.1mol/L KBrO3-KBr 产生的溴加成后,用碘量法测出富马酸的含量,再用0.1mol/L NaOH测出总酸消耗NaOH的毫升数, 由测出的富马酸含量可得其消耗NaOH的毫升数,由总酸消耗NaOH的毫升数减去富马酸消耗NaOH 的毫升数,即可算出2,3二溴丁二酸含量。实验步骤如下:
(1)富马酸的测定
称取0.5000g的2,3二溴丁二酸中间体样品于250mL碘量瓶中,用少许乙醇溶解,加10mL蒸馏水,再加入0.1mol/L KBrO3-KBr溶液和 5mL浓HCl,密封摇匀,以少许15%KI液封口,放置 5min后,加15mL 15%KI溶液,以水封口,再放置 5min,然后加入30mL蒸馏水,以0.1mol/L Na2S2O3溶液滴定近终点时,加入1mL淀粉指示剂,滴至蓝色消失,同时作一空白试验。
计算:含量%=(V空-V 样)x Mx0.116/(Wx2);V空:空白样品消耗Na 2S2O3的毫升数;V样:样品消耗Na 2S 2O 3的毫升数;W:样品重量(克); 0.116:富马酸毫克分子量;M:硫代硫酸钠的摩尔浓度,mol/L。
(2)总酸测定
称取0.2000g的2,3二溴丁二酸 间体样品于250mL碘量瓶中,用少许乙醇溶解,加 30mL蒸馏水,2-3滴酚酞指示剂,用0.1mol/L NaOH 滴至粉红色,记下总消耗氢氧化钠的毫升数。
该方法简便,易于操作,定量准确,可用于生物素合成过程中间体含量的监控。
2. 应用:
2.1 合成柔性羧酸构筑的具有孔洞结构的配合物
马科芳等人通过对反应体系酸碱度的微调节,合成得到了{[Cu(C4H 2Br2O4)] 1/2[Cu(C4H2Br2O4)(H2O)2]1/2(C6H12N4)(H2O)3}n(1);{[Ni(C4H2Br2O4)] 1/2[Ni(C4H2Br2O4)(H2O)2]1/2(C6H12N4)(H2O)3} n(2); [Ni(C 4H 2Br 2O 4)(C3H3N2)2(H 2O) 2] n(3); [Co(C4H2Br2O4)(C3H3N2)2(H2O)2] n(4); [Zn(C4H2Br2O4)(C3H 3N2)2] n(5); [Cd2(C4H2Br2O4)2(C3H3N2)4(H2O)4] n(6)等六个以2, 3-二溴丁二酸为主要骨架构筑的孔洞配合物。举例配合物({Cu2(C4H2Br 2O4)((C6H12N4)(H2O)4}n(1)的合成
取配体2, 3-二溴丁二酸(0.5mmol, 0.136g)溶于15ml甲醇和水的混合溶液中(2: 1),在此溶液中加入Cu(ClO4)2(0.5mmol, 0.124g),搅拌半小时得到一绿色溶液,在此基础上,逐步滴入5ml的六次甲基四胺(1mmol, 0.140g)水溶液,再搅拌半小时,最后用5%的NaOH调节pH到6.0,过滤静置,两天后得到淡绿色晶体。(产率: 80%)。
2. 合成2,3-二溴丁二酸配位聚合物
翁婉丹等人以meso-2,3-二溴丁二酸(H2DBrS)为原料所形成的10例化合物。下表中列出其重要晶体学参数。
物质1-5是Co(II)-Phenanthroline-二溴丁二酸配合物的研究体系中合成的化合物,配合物6-8是二溴丁二酸H2DBr S与Cu(II)构筑的配合物,9、10是Co(II)/Cu(II)与二溴丁二酸、biby构筑的配合物。列举(Hphen)2[(H 2DBr S)(HDBrS)2] 1 的合成:
称取H2DBrS 0.407 g (1.5 mmol) 溶于10 mL MeOH和10 mL H2O的混合溶剂中,在搅拌下缓慢加phen 0.196 g (1.0 mmol)至上述溶液中,产生白色浑浊。过滤浑浊液,分离得到白色沉淀和无色滤液(pH = 2.04),滤液室温下静置培养,数天后发现有无色片状晶体析出。另称取经干燥后的白色沉淀0.1 g 溶于15 mL MeOH 中进行重结晶,数天后发现有无色片状晶体析出。产率83%(以H2DBrS计)。
参考文献:
[1]翁婉丹. 新型2,3-二溴丁二酸配位聚合物的合成、结构及性质[D]. 宁波大学, 2015.
[2]周亚新. 一种快速测定生物素合成中间体2,3二溴丁二酸的方法 [J]. 福建分析测试, 2010, 19 (03): 36-38.
[3]马科芳. 柔性羧酸构筑的具有孔洞结构的配合物的合成、表征及其性质研究[D]. 温州大学, 2010.
1
本文将介绍关于3,5-二氟苯甲酸的微波光谱和理论研究进展,旨在为3,5-二氟苯甲酸的应用提供参考思路。
背景:3,5-二氟苯甲酸(3,5-DFBA)可用于合成药物、染料和农药等多种化合物。此外,它还具有抗菌和抗氧化性质,在一些抗菌和防腐剂中也有广泛应用。苯甲酸是一种平面分子,在各行各业都有广泛的应用,在取代氟原子时会发生重大转变,导致其化学行为和构象偏好发生变化。特别是,氟化苯甲酸通常具有除草和杀真菌特性,这增强了它们的工业相关性。尽管存在用作除草剂的卤取代苯甲酸的毒性数据,但在分子建模中,全面的构象分析仍然是必不可少的,以简化化合物的活性筛选。
3,5-二氟苯甲酸的微波光谱和理论研究:
之前已经有一些关于3,5-DFBA结构的研究。Potrzebowski和Chruszcz报道了它的晶体结构。Dubey等报道了使用晶体结构预测(CSP)对3,5-DFBA的晶体结构研究。傅里叶变换微波谱(FTMW)已被广泛用于探测气相中小分子及其非共价结合配合物的分子结构和动力学。近年来,它已被应用于具有重要环境价值的分子和配合物的研究。Onda等报道了他们使用斯塔克调制吸收光谱(30–40 GHz)和分子束FTMW光谱(6–10 GHz)对苯甲酸进行微波光谱研究。Stark调制光谱的旋转常数报告值与FTMW光谱的旋转常数值不同,特别是旋转常数A。Godfrey和McNaughton也报道了他们使用计算化学和毫米波自由射流斯塔克调制微波光谱(48–72 GHz)对苯甲酸的结构研究,以解决这一差异。
Alitza Gracia等人报告了对 3,5-二氟苯甲酸的理论和光谱综合研究。使用啁啾脉冲傅里叶变换微波 (CP-FTMW) 光谱仪,记录并分析了频率范围为 6 - 12.5 GHz 的旋转光谱。采用量子化学计算来分析 3,5-二氟苯甲酸的构象变化和景观。这些计算重点研究 B3LYP/6-311G 能级沿 CCC=O 和 O=COH 二面角的势能面。方法如下:
利用德克萨斯高级计算中心(Texas Advanced Computing Center, TACC)的集群计算机,通过Gaussian 16程序[21]进行DFT计算,确定氟取代苯甲酸的构象和结构参数。B3LYP (Becke’s三参数混合泛函,使用LYP相关泛函)方法采用6-311G基集,以10?步扫描C-C-C=O和O= C-0-H的二面角,共36步,以识别稳定构象。对O=C的二面角的附加扫描。
宽带光谱是用复旦大学的CP-FTMW光谱仪测量的,在之前的两篇出版物的补充部分进行了描述。该光谱仪的设计与先前报道的其他啁啾脉冲宽带光谱仪相似。使用商业样品3,5-二氟苯甲酸(来自Adamas-beta),无需进一步纯化。3,5-二氟苯甲酸样品被放置在一个储液器中,该储液器是定制的脉冲阀(通用阀系列9)的一部分,靠近阀孔并加热到200℃。在约4x105 Pa的停滞压力下,氖气作为载气产生分子束。然后将气体混合物膨胀到真空室(~5x10- 4pa),产生脉冲(7Hz)超音速射流。延时960us后,由25 GS/s任意波形发生器(AWG)产生10个连续啁啾脉冲,持续时间为4μs,范围为6-12.5GHz。这些脉冲随后被一个200瓦的固态放大器放大,并通过喇叭天线广播到真空室。微波信号与分子束在相互垂直的方向上相互作用。从另一端的喇叭天线采集分子系宏观随后的十次自由感应衰减(FID)信号,经过保护限幅器、开关和低噪声放大器,最后记录在数字示波器上。频谱测频精度达到15kHz,分辨率优于25kHz。采集500k时域FID信号,通过快速傅里叶变换得到6 ~ 12.5GHz频段的宽带频谱。
结果:



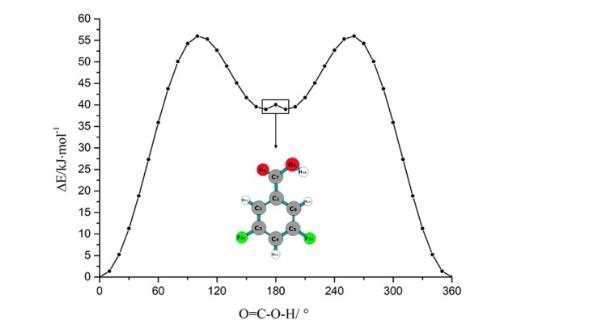
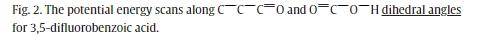
在这项工作中,研究人员首次对3,5-二氟苯甲酸进行了理论和高分辨率光谱研究。通过量子化学计算,确定了3,5-二氟苯甲酸的两种构象。经零点能量修正后的构象能差为26.5 kJ/mol。该分子在其全局最小构象中倾向于平面结构。第二个构象可能由于不利的氢-氢相互作用而包含双最小电位面。实验确定了3,5-二氟苯甲酸的最小能量构象。用半刚性转子模型哈密顿量分析了母同位素和7个13C取代同位素的谱。测定了高精度的旋转常数。利用这些同位素物的旋转常数推导了取代结构。理论与实验在转动常数上的一致性非常好。
参考文献:
[1]Gracia A, Hong J, Arismendi R, et al. Conformational landscapes of symmetrically fluorine-substituted benzoic acids I: Microwave spectroscopic and theoretical studies on 3, 5-difluorobenzoic acid[J]. Journal of Molecular Spectroscopy, 2023, 397: 111839.
显示全部本文将介绍关于3,5-二氟苯甲酸的微波光谱和理论研究进展,旨在为3,5-二氟苯甲酸的应用提供参考思路。
背景:3,5-二氟苯甲酸(3,5-DFBA)可用于合成药物、染料和农药等多种化合物。此外,它还具有抗菌和抗氧化性质,在一些抗菌和防腐剂中也有广泛应用。苯甲酸是一种平面分子,在各行各业都有广泛的应用,在取代氟原子时会发生重大转变,导致其化学行为和构象偏好发生变化。特别是,氟化苯甲酸通常具有除草和杀真菌特性,这增强了它们的工业相关性。尽管存在用作除草剂的卤取代苯甲酸的毒性数据,但在分子建模中,全面的构象分析仍然是必不可少的,以简化化合物的活性筛选。
3,5-二氟苯甲酸的微波光谱和理论研究:
之前已经有一些关于3,5-DFBA结构的研究。Potrzebowski和Chruszcz报道了它的晶体结构。Dubey等报道了使用晶体结构预测(CSP)对3,5-DFBA的晶体结构研究。傅里叶变换微波谱(FTMW)已被广泛用于探测气相中小分子及其非共价结合配合物的分子结构和动力学。近年来,它已被应用于具有重要环境价值的分子和配合物的研究。Onda等报道了他们使用斯塔克调制吸收光谱(30–40 GHz)和分子束FTMW光谱(6–10 GHz)对苯甲酸进行微波光谱研究。Stark调制光谱的旋转常数报告值与FTMW光谱的旋转常数值不同,特别是旋转常数A。Godfrey和McNaughton也报道了他们使用计算化学和毫米波自由射流斯塔克调制微波光谱(48–72 GHz)对苯甲酸的结构研究,以解决这一差异。
Alitza Gracia等人报告了对 3,5-二氟苯甲酸的理论和光谱综合研究。使用啁啾脉冲傅里叶变换微波 (CP-FTMW) 光谱仪,记录并分析了频率范围为 6 - 12.5 GHz 的旋转光谱。采用量子化学计算来分析 3,5-二氟苯甲酸的构象变化和景观。这些计算重点研究 B3LYP/6-311G 能级沿 CCC=O 和 O=COH 二面角的势能面。方法如下:
利用德克萨斯高级计算中心(Texas Advanced Computing Center, TACC)的集群计算机,通过Gaussian 16程序[21]进行DFT计算,确定氟取代苯甲酸的构象和结构参数。B3LYP (Becke’s三参数混合泛函,使用LYP相关泛函)方法采用6-311G基集,以10?步扫描C-C-C=O和O= C-0-H的二面角,共36步,以识别稳定构象。对O=C的二面角的附加扫描。
宽带光谱是用复旦大学的CP-FTMW光谱仪测量的,在之前的两篇出版物的补充部分进行了描述。该光谱仪的设计与先前报道的其他啁啾脉冲宽带光谱仪相似。使用商业样品3,5-二氟苯甲酸(来自Adamas-beta),无需进一步纯化。3,5-二氟苯甲酸样品被放置在一个储液器中,该储液器是定制的脉冲阀(通用阀系列9)的一部分,靠近阀孔并加热到200℃。在约4x105 Pa的停滞压力下,氖气作为载气产生分子束。然后将气体混合物膨胀到真空室(~5x10- 4pa),产生脉冲(7Hz)超音速射流。延时960us后,由25 GS/s任意波形发生器(AWG)产生10个连续啁啾脉冲,持续时间为4μs,范围为6-12.5GHz。这些脉冲随后被一个200瓦的固态放大器放大,并通过喇叭天线广播到真空室。微波信号与分子束在相互垂直的方向上相互作用。从另一端的喇叭天线采集分子系宏观随后的十次自由感应衰减(FID)信号,经过保护限幅器、开关和低噪声放大器,最后记录在数字示波器上。频谱测频精度达到15kHz,分辨率优于25kHz。采集500k时域FID信号,通过快速傅里叶变换得到6 ~ 12.5GHz频段的宽带频谱。
结果:
在这项工作中,研究人员首次对3,5-二氟苯甲酸进行了理论和高分辨率光谱研究。通过量子化学计算,确定了3,5-二氟苯甲酸的两种构象。经零点能量修正后的构象能差为26.5 kJ/mol。该分子在其全局最小构象中倾向于平面结构。第二个构象可能由于不利的氢-氢相互作用而包含双最小电位面。实验确定了3,5-二氟苯甲酸的最小能量构象。用半刚性转子模型哈密顿量分析了母同位素和7个13C取代同位素的谱。测定了高精度的旋转常数。利用这些同位素物的旋转常数推导了取代结构。理论与实验在转动常数上的一致性非常好。
参考文献:
[1]Gracia A, Hong J, Arismendi R, et al. Conformational landscapes of symmetrically fluorine-substituted benzoic acids I: Microwave spectroscopic and theoretical studies on 3, 5-difluorobenzoic acid[J]. Journal of Molecular Spectroscopy, 2023, 397: 111839.
1
本文将讲述5-溴-4-氯-3-吲哚磷酸如何用于酸性磷酸酯酶活性测定,旨在为其在临床应用提供参考。
简述:5-溴-4-氯-3-吲哚磷酸对甲苯胺盐,英文名称:5-Bromo-4-chloro-3-indolyl phosphate p-toluidine salt,CAS:6578-06-9,分子式:C15H15BrClN2O4P,外观与性状:片状晶体。5-溴-4-氯-3-吲哚磷酸对甲苯胺盐是显色底物,用于碱性磷酸酶活性的比色法检测。
应用:用于酸性磷酸酯酶活性测定。
在外加交变磁场中,磁性膜片受磁场激发产生磁矩,将磁能转换为机械能,并产生沿长度方向伸 缩振动,即磁致伸缩。当交变磁场频率与磁性膜片机械振动频率相等时,膜片产生共振,此时具有最大振幅,此时振动频率为磁性膜片共振频率。当磁性膜片传感器表面负载质量发生变化时,其共振频率也会随之改变。由于磁性膜片本身是磁性的,其伸缩振动产生磁通,产生的磁通可由检测线圈检测。磁弹性传感器中信号的激发与传送是通过磁场进行的,传感器与检测仪器之间没有任何物理连接,属于无线无源传感器,磁弹性传感器这一特点使得它在活体、在体分析中具有广泛的应用前景。
吴仕辉等人提出了一种新型磁弹性传感器用于酸性磷酸酯酶活性检测,该方法易操作,所用试剂简单,不需其它辅助试剂,且检测装置简易,便宜。该方法检测原理是以5-溴-4-氯-3-吲哚-磷酸对甲苯胺盐(BCIP)为底物,经酸性磷酸酯酶水解后生成兰色二聚物沉淀,该二聚物沉淀能紧密附着在传感器表面,导致传感器共振频率发生变化,传感器共振频率变化幅度正比于酸性磷酸酯酶浓度。具体如下:
(1)传感器修饰
第一步,先用超声波清洗传感器,再用水和乙醇清洗,后用氮气吹干; 第二步,将10 μl Bayhydrol 110均匀涂在传感器表面,在空气中晾干后置于 150℃ 烘箱中烘2小时,使传感器表面形成一层牢固的保护膜,以保护磁片不被腐蚀的作用;第三步,将含0.02% EDC和NHS的BSA溶液涂在传感器表面,在空气中干燥;第四步,将干燥后的传感器置于冰箱保存。
(2)检测
将上述修饰后的传感器置于含1.5 mg/ml BCIP的柠檬酸缓冲溶液中(pH 4.8),在37℃下恒温10 min,使得传感器响应趋于稳定,并记录传感器此时共振频率?0。 然后将准备好的1.5~30 U/l系列酸性磷酸酯酶溶液加入至该反应池中开始反应。随 着反应时间进行,不断有沉淀附着在传感器表面,传感器共振频率发生改变,??=?-?0。实验中通过记录传感器在反应时间100 min内共振频率的变化幅度(??)来分析酸性磷酸酯酶的活性。所有实验数据均为三次平行测定后的平均值。
(3)酸性磷酸酯酶的测定
以5-溴-4-氯-3-吲哚-磷酸对甲苯胺盐(BCIP)为底物的酸性磷酸酯酶催化水解反应的示意图如下图所示,酸性磷酸酯酶水解BCIP脱磷酸后得一中间产物,该中间产物在与溶液中的溶解氧的作用下迅速异构及二聚为一种兰色二聚物沉淀。这种兰色二聚物能紧密的附着在传感器表面,导致传感器的共振频率发生改变,且灵敏度较好。
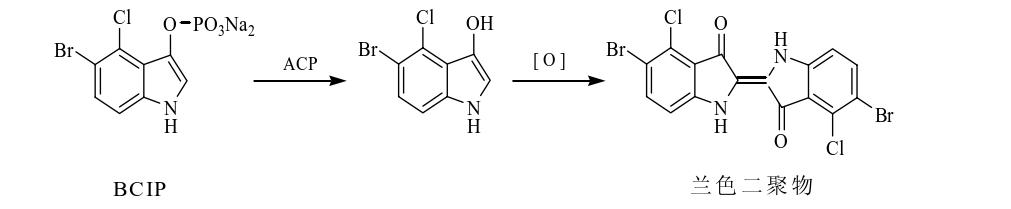
(4)总结
实验中通过记录传感器共振频率变化幅度来检测酸性磷酸酯酶活性。同时还考察底物浓度对酶催化反应的影响,并通过BSA在传感器表面的固定对所生成的兰色二聚物在传感器表面附着能力进行了改善。该方法可用来检测1.5~15 U/l酸性磷酸酯酶,检测限达到1.5 U/l,检验灵敏度与分光光度法相当。由于该传感器制作简单,响应灵敏,并能实现无线检测,在临床应用中具有广泛的应用前景。
参考文献:
[1] 吴仕辉. 磁弹性无线生物传感器在生化分析中的应用研究[D]. 湖南:湖南大学,2006. DOI:10.7666/d.y892959.
显示全部本文将讲述5-溴-4-氯-3-吲哚磷酸如何用于酸性磷酸酯酶活性测定,旨在为其在临床应用提供参考。
简述:5-溴-4-氯-3-吲哚磷酸对甲苯胺盐,英文名称:5-Bromo-4-chloro-3-indolyl phosphate p-toluidine salt,CAS:6578-06-9,分子式:C15H15BrClN2O4P,外观与性状:片状晶体。5-溴-4-氯-3-吲哚磷酸对甲苯胺盐是显色底物,用于碱性磷酸酶活性的比色法检测。
应用:用于酸性磷酸酯酶活性测定。
在外加交变磁场中,磁性膜片受磁场激发产生磁矩,将磁能转换为机械能,并产生沿长度方向伸 缩振动,即磁致伸缩。当交变磁场频率与磁性膜片机械振动频率相等时,膜片产生共振,此时具有最大振幅,此时振动频率为磁性膜片共振频率。当磁性膜片传感器表面负载质量发生变化时,其共振频率也会随之改变。由于磁性膜片本身是磁性的,其伸缩振动产生磁通,产生的磁通可由检测线圈检测。磁弹性传感器中信号的激发与传送是通过磁场进行的,传感器与检测仪器之间没有任何物理连接,属于无线无源传感器,磁弹性传感器这一特点使得它在活体、在体分析中具有广泛的应用前景。
吴仕辉等人提出了一种新型磁弹性传感器用于酸性磷酸酯酶活性检测,该方法易操作,所用试剂简单,不需其它辅助试剂,且检测装置简易,便宜。该方法检测原理是以5-溴-4-氯-3-吲哚-磷酸对甲苯胺盐(BCIP)为底物,经酸性磷酸酯酶水解后生成兰色二聚物沉淀,该二聚物沉淀能紧密附着在传感器表面,导致传感器共振频率发生变化,传感器共振频率变化幅度正比于酸性磷酸酯酶浓度。具体如下:
(1)传感器修饰
第一步,先用超声波清洗传感器,再用水和乙醇清洗,后用氮气吹干; 第二步,将10 μl Bayhydrol 110均匀涂在传感器表面,在空气中晾干后置于 150℃ 烘箱中烘2小时,使传感器表面形成一层牢固的保护膜,以保护磁片不被腐蚀的作用;第三步,将含0.02% EDC和NHS的BSA溶液涂在传感器表面,在空气中干燥;第四步,将干燥后的传感器置于冰箱保存。
(2)检测
将上述修饰后的传感器置于含1.5 mg/ml BCIP的柠檬酸缓冲溶液中(pH 4.8),在37℃下恒温10 min,使得传感器响应趋于稳定,并记录传感器此时共振频率?0。 然后将准备好的1.5~30 U/l系列酸性磷酸酯酶溶液加入至该反应池中开始反应。随 着反应时间进行,不断有沉淀附着在传感器表面,传感器共振频率发生改变,??=?-?0。实验中通过记录传感器在反应时间100 min内共振频率的变化幅度(??)来分析酸性磷酸酯酶的活性。所有实验数据均为三次平行测定后的平均值。
(3)酸性磷酸酯酶的测定
以5-溴-4-氯-3-吲哚-磷酸对甲苯胺盐(BCIP)为底物的酸性磷酸酯酶催化水解反应的示意图如下图所示,酸性磷酸酯酶水解BCIP脱磷酸后得一中间产物,该中间产物在与溶液中的溶解氧的作用下迅速异构及二聚为一种兰色二聚物沉淀。这种兰色二聚物能紧密的附着在传感器表面,导致传感器的共振频率发生改变,且灵敏度较好。
(4)总结
实验中通过记录传感器共振频率变化幅度来检测酸性磷酸酯酶活性。同时还考察底物浓度对酶催化反应的影响,并通过BSA在传感器表面的固定对所生成的兰色二聚物在传感器表面附着能力进行了改善。该方法可用来检测1.5~15 U/l酸性磷酸酯酶,检测限达到1.5 U/l,检验灵敏度与分光光度法相当。由于该传感器制作简单,响应灵敏,并能实现无线检测,在临床应用中具有广泛的应用前景。
参考文献:
[1] 吴仕辉. 磁弹性无线生物传感器在生化分析中的应用研究[D]. 湖南:湖南大学,2006. DOI:10.7666/d.y892959.
1
通过对对叔丁基氯苄的分析和合成过程的探索,为科研人员和相关研究人员提供了有关该化合物的重要信息,旨在为未来的研究和开发工作提供了指导。
背景:对叔丁基氯苄是一种用途十分广泛的精细化工中间体,在香料行业用于生产铃兰醛,农药行业生产吡螨胺和哒螨酮等,医药行业生产安其敏和布替萘芬等等。随着精细化工新产品的不断开发,其应用范围不断扩展用量不断增加,对它的质量也有更高要求。
1. 合成:
(1)方法一:
通过采用特定氯化促进剂A替代氯化氢通入的方法 ,以叔丁基苯为原料进行氯甲基化制备对叔丁基氯苄 ,使目标产物的平均含量和收率达到 97%以上。反应母液经分离后循环使用仍保持相当活性 ,对产品纯度和收率无不利影响。具体步骤如下:
向1000mL三口瓶中依次加入20g多聚甲醛、64g浓盐酸、150g 36%甲醛溶液、60g氯化锌配制成母液, 然后加入200g叔丁基苯, 充分混合, 加热至80℃左右, 于10h内缓慢滴加约180g氯化促进剂A。用气相色谱跟踪检测有机层, 反应液中无原料叔丁基苯后, 冷却反应液、静置分出有机层, 然后用水洗涤, 去除其中的水溶物, 干燥后得到产品。分出的含醛母液经分析, 补充甲醛后即可循环用于下次合成。
(2)方法二:
以叔丁基苯、多聚甲醛和盐酸为原料,通过氯甲基化反应制备对叔丁基氯苄。最佳工艺条件为:磷酸作催化剂,叔丁基苯∶甲醛∶氯化氢=1∶3∶3(mol/mol),反应温度80~85℃,反应时间15h。具体实验操作如下:
500 ml三口瓶中,依次加入53.6 g(0.40 mol)叔丁基苯,22.0 g(按甲醛计算为0.73 mol)多聚甲醛,60.0 ml冰醛酸,40.0 ml 85%磷酸,80.0 ml(0.85 mol)36%盐酸,剧烈搅拌下,水浴加热至80 ℃,控制反应温度80~85 ℃,反应15 h。冷却至室温,分出粗产品,依次水洗(50 ml×2),1 0%K2CO3洗(50 ml×1),水洗(50 ml×2),加入20 ml乙醚,无水K2CO3干燥过夜。先常压蒸去乙醚,然后减压蒸馏,前馏分45~75 ℃/267 Pa,回收叔丁基苯30.4 g, 转化率43.2%(以加入的叔丁基苯计算,下同);正馏分80~82 ℃/267 Pa,得产物对叔丁基氯苄20.4 g, 收率64.7%。
2. 分析:
曹俭等人建立了叔丁基氯苄的反相高效液相色谱分析方法。实验方法为:流动相为甲醇/水 (80/20, V/V) , 流速1.0mL/min, 紫外检测波长230nm, 柱温25℃, 进样量20μL (以下进样量均为20μL) 。用外标法定量, 标样和样品均用甲醇溶解, 每个样品重复进样3次, 以峰面积为计算标准。样品测定的色谱图如图所示, 图中8.519min处最大峰即为叔丁基氯苄峰。方法的相对标准偏差为0.36%,平均回收率为100.8%。

参考文献:
[1]曹俭,肖稳发,胡顺忠. 高效液相色谱分析叔丁基氯苄含量 [J]. 光谱实验室, 2010, 27 (01): 280-282.
[2]王大威,左华,沙磊等. 对叔丁基氯苄的合成工艺研究 [J]. 精细化工中间体, 2003, (04): 24-25+55. DOI:10.19342/j.cnki.issn.1009-9212.2003.04.010.
[3]杨建瑜. 对叔丁基氯苄的合成新方法 [J]. 四川化工与腐蚀控制, 2003, (05): 1-4.
显示全部通过对对叔丁基氯苄的分析和合成过程的探索,为科研人员和相关研究人员提供了有关该化合物的重要信息,旨在为未来的研究和开发工作提供了指导。
背景:对叔丁基氯苄是一种用途十分广泛的精细化工中间体,在香料行业用于生产铃兰醛,农药行业生产吡螨胺和哒螨酮等,医药行业生产安其敏和布替萘芬等等。随着精细化工新产品的不断开发,其应用范围不断扩展用量不断增加,对它的质量也有更高要求。
1. 合成:
(1)方法一:
通过采用特定氯化促进剂A替代氯化氢通入的方法 ,以叔丁基苯为原料进行氯甲基化制备对叔丁基氯苄 ,使目标产物的平均含量和收率达到 97%以上。反应母液经分离后循环使用仍保持相当活性 ,对产品纯度和收率无不利影响。具体步骤如下:
向1000mL三口瓶中依次加入20g多聚甲醛、64g浓盐酸、150g 36%甲醛溶液、60g氯化锌配制成母液, 然后加入200g叔丁基苯, 充分混合, 加热至80℃左右, 于10h内缓慢滴加约180g氯化促进剂A。用气相色谱跟踪检测有机层, 反应液中无原料叔丁基苯后, 冷却反应液、静置分出有机层, 然后用水洗涤, 去除其中的水溶物, 干燥后得到产品。分出的含醛母液经分析, 补充甲醛后即可循环用于下次合成。
(2)方法二:
以叔丁基苯、多聚甲醛和盐酸为原料,通过氯甲基化反应制备对叔丁基氯苄。最佳工艺条件为:磷酸作催化剂,叔丁基苯∶甲醛∶氯化氢=1∶3∶3(mol/mol),反应温度80~85℃,反应时间15h。具体实验操作如下:
500 ml三口瓶中,依次加入53.6 g(0.40 mol)叔丁基苯,22.0 g(按甲醛计算为0.73 mol)多聚甲醛,60.0 ml冰醛酸,40.0 ml 85%磷酸,80.0 ml(0.85 mol)36%盐酸,剧烈搅拌下,水浴加热至80 ℃,控制反应温度80~85 ℃,反应15 h。冷却至室温,分出粗产品,依次水洗(50 ml×2),1 0%K2CO3洗(50 ml×1),水洗(50 ml×2),加入20 ml乙醚,无水K2CO3干燥过夜。先常压蒸去乙醚,然后减压蒸馏,前馏分45~75 ℃/267 Pa,回收叔丁基苯30.4 g, 转化率43.2%(以加入的叔丁基苯计算,下同);正馏分80~82 ℃/267 Pa,得产物对叔丁基氯苄20.4 g, 收率64.7%。
2. 分析:
曹俭等人建立了叔丁基氯苄的反相高效液相色谱分析方法。实验方法为:流动相为甲醇/水 (80/20, V/V) , 流速1.0mL/min, 紫外检测波长230nm, 柱温25℃, 进样量20μL (以下进样量均为20μL) 。用外标法定量, 标样和样品均用甲醇溶解, 每个样品重复进样3次, 以峰面积为计算标准。样品测定的色谱图如图所示, 图中8.519min处最大峰即为叔丁基氯苄峰。方法的相对标准偏差为0.36%,平均回收率为100.8%。
参考文献:
[1]曹俭,肖稳发,胡顺忠. 高效液相色谱分析叔丁基氯苄含量 [J]. 光谱实验室, 2010, 27 (01): 280-282.
[2]王大威,左华,沙磊等. 对叔丁基氯苄的合成工艺研究 [J]. 精细化工中间体, 2003, (04): 24-25+55. DOI:10.19342/j.cnki.issn.1009-9212.2003.04.010.
[3]杨建瑜. 对叔丁基氯苄的合成新方法 [J]. 四川化工与腐蚀控制, 2003, (05): 1-4.
1
这篇文章将详细介绍如何利用4-硝基邻苯二甲酸合成金属配合物,这一方法在有机合成和材料等领域具有重要的应用和意义。
简述:4-硝基邻苯二甲酸,英文名称:4-Nitrophthalic acid,CAS:610-27-5,分子式:C8H5NO6,外观与性状:米色固体。4-硝基邻苯二甲酸常用作有机合成中间体,可用于合成各种金属配合物。
应用:合成金属配合物
金属有机骨架(MOFs)因为具有有趣的分子构架和拓扑结构,而且其可作为功能材料在气体吸附、分离、多相催化、光致发光等许多领域有巨大 应用潜力,因此,已成为一个新的研究热点.芳香族苯二甲酸及其衍生物(如1,n-苯二甲酸,n =2,3,4)由于其具有令人感兴趣的结构和性质,在配位聚合物自组装中作为构筑模块而得到广泛应用。在羧基中存在协同配位的氧原子,使得其有多种配位模式(单齿配位,双齿桥联和鳌合),这也有助于拓扑结构的多样化。
1. 合成金属锰配位聚合物{[Mn(4-Nbdc)(bib)1.5(H2O)]·0.5H2O}n
尹卫东等人以4-硝基邻苯二甲酸为主配体,1,4-二(1-咪唑基)苯为辅助配体,通过与醋酸锰反应合成了金属锰配位聚合物{[Mn(4-Nbdc)(bib)1.5(H2O)]·0.5H2O}n。经过4-硝基邻苯二甲酸和辅助配体1,4-二(1-咪唑基)苯与金属锰配位形成一维阶梯链状结构聚合物,并通过氢键和π…π作用进一步形成完整的三维超分子结构。配合物的合成步骤如下:
将Mn(OAc)2·4H2O(24.5 mg,0.10 mmol),4-硝基邻苯二甲酸(21.2 mg,0.1 mmol)、1,4-二(1-咪唑基)苯(42.0 mg,0.20 mmol)和水(7 mL)放置在一个23 mL的不锈钢反应釜内,在393 K下反应4天,然后冷却至室温,得到产率约86%的无色块状晶体。经元素分析得该化合物的化学式为C26H22N7O8Mn,C、H、N元素含量的计算值分别为(%):C:50.74;H:3.60;N:15.93;理论值分别为(%):C:50.59;H:3.64;N:15.90。
2. 合成4-硝基邻苯二甲酸Cu(Ⅱ)聚合物∞1{[Cu(Him)(H2O)L]·0.5H2O}
周林霞等人以4-硝基邻苯二甲酸(H2L)为有机配体,咪唑(Him)为第二配体合成了结构新颖的一维链状Cu聚合物:1∞{[Cu(Him)(H2O)L]·0.5H2O}。结构单元中,该链状Cu聚合物首先通过O—H…O氢键形成一维双链和二维层,层与层之间进一步相互穿插,通过芳环堆积作用形成三维超分子网络结构。配合物的合成步骤如下:
滴加2.0 mL 1 mol/L NaOH至经适量蒸馏水溶解的0.890 g(0.5 mmol)CuCl2·2H2O溶液,形成墨绿色Cu(OH)2·x H2O沉淀,该沉淀经5次离心分离、蒸馏水洗涤后备用。搅拌下,将0.106 g 4-硝基邻苯二甲酸溶于10 mL H2O和10 mL EtOH组成的混合溶剂中,再加入备用的 Cu(OH)2·x H2O沉淀,形成浅绿色悬浊液,继续搅拌15 min后,再加入0.035 g(0.5 mmol)咪唑固体,咪唑溶解,颜色加深,溶液几近澄清,又搅拌 10 min后过滤,滤液静置,测定其pH为3.73,约2个月后发现有少量蓝色晶体1∞{[Cu(Him)(H2O)L]·0.5H2O}混在一些蓝色粉末中。
3. 合成铜配位聚合物
付立海等人在水热条件下以4-硝基邻苯二甲酸(4-H2nph)/2,2'-联吡啶-4,4'-二甲酸(H2bda)为主要配体,3-(2-吡啶基)吡唑(HL)/1,4-双(咪唑基-1-基)丁烷(bib)为辅助配体制备了 2种新的金属有机配位聚合物[Cu4(4-nph)(L)6]n(1)和{[Cu2(bda)2(bib)2(H2O)4]·4H2O}n(2)。在配合物1中,羧基配体4-nph2-通过单齿模式桥联金属中心,形成一维结构。此外,还用Gaussian16程序PBEO/LANL2DZ方法从配合物1的晶体结构中提取"分子片段"进行了量子化学计算.计算结果表明配位原子与铜(Ⅱ)离子之间存在着共价作用。
参考文献:
[1]付立海,李秀梅,刘博等. 两种铜配位聚合物的晶体结构和量子化学计算(英文) [J]. 无机化学学报, 2022, 38 (11): 2249-2258.
[2]尹卫东,何钰莹,沈佳. 金属锰配位聚合物{[Mn(4-Nbdc)(bib)_(1.5)(H_2O)]·0.5H_2O}_n的合成与晶体结构 [J]. 洛阳师范学院学报, 2017, 36 (11): 40-43. DOI:10.16594/j.cnki.41-1302/g4.2017.11.012.
[3]周林霞. 4-硝基邻苯二甲酸Cu(Ⅱ)聚合物_∞~1{[Cu(Him)(H_2O)L]·0.5H_2O}的合成与晶体结构表征 [J]. 化学试剂, 2014, 36 (07): 663-666+669. DOI:10.13822/j.cnki.hxsj.2014.07.021.
显示全部这篇文章将详细介绍如何利用4-硝基邻苯二甲酸合成金属配合物,这一方法在有机合成和材料等领域具有重要的应用和意义。
简述:4-硝基邻苯二甲酸,英文名称:4-Nitrophthalic acid,CAS:610-27-5,分子式:C8H5NO6,外观与性状:米色固体。4-硝基邻苯二甲酸常用作有机合成中间体,可用于合成各种金属配合物。
应用:合成金属配合物
金属有机骨架(MOFs)因为具有有趣的分子构架和拓扑结构,而且其可作为功能材料在气体吸附、分离、多相催化、光致发光等许多领域有巨大 应用潜力,因此,已成为一个新的研究热点.芳香族苯二甲酸及其衍生物(如1,n-苯二甲酸,n =2,3,4)由于其具有令人感兴趣的结构和性质,在配位聚合物自组装中作为构筑模块而得到广泛应用。在羧基中存在协同配位的氧原子,使得其有多种配位模式(单齿配位,双齿桥联和鳌合),这也有助于拓扑结构的多样化。
1. 合成金属锰配位聚合物{[Mn(4-Nbdc)(bib)1.5(H2O)]·0.5H2O}n
尹卫东等人以4-硝基邻苯二甲酸为主配体,1,4-二(1-咪唑基)苯为辅助配体,通过与醋酸锰反应合成了金属锰配位聚合物{[Mn(4-Nbdc)(bib)1.5(H2O)]·0.5H2O}n。经过4-硝基邻苯二甲酸和辅助配体1,4-二(1-咪唑基)苯与金属锰配位形成一维阶梯链状结构聚合物,并通过氢键和π…π作用进一步形成完整的三维超分子结构。配合物的合成步骤如下:
将Mn(OAc)2·4H2O(24.5 mg,0.10 mmol),4-硝基邻苯二甲酸(21.2 mg,0.1 mmol)、1,4-二(1-咪唑基)苯(42.0 mg,0.20 mmol)和水(7 mL)放置在一个23 mL的不锈钢反应釜内,在393 K下反应4天,然后冷却至室温,得到产率约86%的无色块状晶体。经元素分析得该化合物的化学式为C26H22N7O8Mn,C、H、N元素含量的计算值分别为(%):C:50.74;H:3.60;N:15.93;理论值分别为(%):C:50.59;H:3.64;N:15.90。
2. 合成4-硝基邻苯二甲酸Cu(Ⅱ)聚合物∞1{[Cu(Him)(H2O)L]·0.5H2O}
周林霞等人以4-硝基邻苯二甲酸(H2L)为有机配体,咪唑(Him)为第二配体合成了结构新颖的一维链状Cu聚合物:1∞{[Cu(Him)(H2O)L]·0.5H2O}。结构单元中,该链状Cu聚合物首先通过O—H…O氢键形成一维双链和二维层,层与层之间进一步相互穿插,通过芳环堆积作用形成三维超分子网络结构。配合物的合成步骤如下:
滴加2.0 mL 1 mol/L NaOH至经适量蒸馏水溶解的0.890 g(0.5 mmol)CuCl2·2H2O溶液,形成墨绿色Cu(OH)2·x H2O沉淀,该沉淀经5次离心分离、蒸馏水洗涤后备用。搅拌下,将0.106 g 4-硝基邻苯二甲酸溶于10 mL H2O和10 mL EtOH组成的混合溶剂中,再加入备用的 Cu(OH)2·x H2O沉淀,形成浅绿色悬浊液,继续搅拌15 min后,再加入0.035 g(0.5 mmol)咪唑固体,咪唑溶解,颜色加深,溶液几近澄清,又搅拌 10 min后过滤,滤液静置,测定其pH为3.73,约2个月后发现有少量蓝色晶体1∞{[Cu(Him)(H2O)L]·0.5H2O}混在一些蓝色粉末中。
3. 合成铜配位聚合物
付立海等人在水热条件下以4-硝基邻苯二甲酸(4-H2nph)/2,2'-联吡啶-4,4'-二甲酸(H2bda)为主要配体,3-(2-吡啶基)吡唑(HL)/1,4-双(咪唑基-1-基)丁烷(bib)为辅助配体制备了 2种新的金属有机配位聚合物[Cu4(4-nph)(L)6]n(1)和{[Cu2(bda)2(bib)2(H2O)4]·4H2O}n(2)。在配合物1中,羧基配体4-nph2-通过单齿模式桥联金属中心,形成一维结构。此外,还用Gaussian16程序PBEO/LANL2DZ方法从配合物1的晶体结构中提取"分子片段"进行了量子化学计算.计算结果表明配位原子与铜(Ⅱ)离子之间存在着共价作用。
参考文献:
[1]付立海,李秀梅,刘博等. 两种铜配位聚合物的晶体结构和量子化学计算(英文) [J]. 无机化学学报, 2022, 38 (11): 2249-2258.
[2]尹卫东,何钰莹,沈佳. 金属锰配位聚合物{[Mn(4-Nbdc)(bib)_(1.5)(H_2O)]·0.5H_2O}_n的合成与晶体结构 [J]. 洛阳师范学院学报, 2017, 36 (11): 40-43. DOI:10.16594/j.cnki.41-1302/g4.2017.11.012.
[3]周林霞. 4-硝基邻苯二甲酸Cu(Ⅱ)聚合物_∞~1{[Cu(Him)(H_2O)L]·0.5H_2O}的合成与晶体结构表征 [J]. 化学试剂, 2014, 36 (07): 663-666+669. DOI:10.13822/j.cnki.hxsj.2014.07.021.
1
2,4-二甲基-6-叔丁基苯酚作为防止甲基丙烯酸甲酯自聚所加的一定量的一种物质,它的定量检测很有必要。
背景:2,4-二甲基-6-叔丁基苯酚是一种能够快速与游离基反应并终止链反应的物质,通常被称为稳定剂。在储存或运输甲基丙烯酸甲酯时,添加适量的该物质可以防止甲基丙烯酸甲酯的自聚。若添加量过少,则无法有效阻止自聚;而添加量过多则会影响后续的聚合反应。因此,建立一种简便、快速、准确测定稳定剂含量的方法对于指导工艺运行具有非常重要的意义。
含量测定:
1. 方法一:
苏桂艳等人利用Rtx-wax毛细管柱,以乙腈为溶剂,氢火焰离子检测器检测,对工业用甲基丙烯酸甲酯中阻聚剂2,4-二甲基- 6-叔丁基苯酚含量进行测定。实验部分如下:
(1)色谱操作条件:
色谱柱:WCOT熔融石英15m×0.53 mm×1.0mm。进样口温度:250℃;检测器温度:250℃;柱温:初始温度100℃,保持 5min,以15℃/min速率加热到220℃,保持5min;分流比:1:10; 载气:氦气,恒压21.5kpa;进样量:1μL。
(2)校准曲线制备
标准储备溶液的配制:准确称取0.05g 2,4-二甲基-6-叔丁基苯酚置于50ml容量瓶中,用乙腈定容。
校验溶液的配制:分别称取0.1g,0.3g、0.5g标准储备溶液,精确至0.1mg,分别加入到50mL容量瓶,乙腈定容至刻度。
(3)试样分析步骤
试样混匀后直接注入色谱仪中测定。
实验结果为:该方法回收率为 98.5~101.5%,相对标准偏差为1.7~2.0%。此方法简便、快速、灵敏度高,并具有较好的准确度和精密度,适用于对甲基丙烯酸甲酯中阻聚剂2,4-二甲基-6-叔丁基苯酚的质控分析。
2. 方法二:
刘兴富等人提出用气相色谱外标法测定甲基丙烯酸甲酯中阻聚剂 2 ,4 -二甲基 - 6 -叔丁基苯酚的质量分数。实验部分如下:
(1)色谱条件:
色谱柱为SE-30填充柱,规格为2 m×3 mm;柱温130 ℃,汽化温度250 ℃;检测器温度250 ℃;柱前压40 kPa;灵敏度(RANGE)102;氢气流速60 mL/min;空气流速500 mL/min。
(2)工作曲线的绘制
精密称取0.010 7 g 2,4-二甲基-6-叔丁基苯酚标准品于100 mL容量瓶中,用甲醇稀释至刻度,摇匀。分别移取上述溶液2.5 mL、5 mL、10 mL、20 mL置于50 mL容量瓶中,用甲醇稀释至刻度,摇匀。待色谱仪稳定后进样,进样量均为2微升。
以峰面积对2,4-二甲基-6-叔丁基苯酚质量作图,绘制工作曲线。见图。

结果:采用气相色谱外标法测定甲基丙烯酸甲酯中阻聚剂2,4-二甲基-6-叔丁基苯酚的含量。在最佳底液条件下,加入一定量标准,对其在24 h内进行多次测定,定量结果的重复性好,且线性关系很好,说明此方法稳定性好。在甲基丙烯酸甲酯中分别加入2,4-二甲基-6-叔丁基苯酚标准,测得的平均回收率在96 %以上,说明该法用于甲基丙烯酸甲酯中的2,4-二甲基-6-叔丁基苯酚测定准确可靠。
参考文献:
[1]苏桂艳,王东川,苏建萍. 甲基丙烯酸甲酯中阻聚剂2,4-二甲基-6-叔丁基苯酚含量的测定 [J]. 化工管理, 2017, (23): 206.
[2]刘兴富,丛文俊,徐金贤. 甲基丙烯酸甲酯中阻聚剂2,4-二甲基-6-叔丁基苯酚的测定 [J]. 辽宁化工, 2004, (07): 432-433.
显示全部2,4-二甲基-6-叔丁基苯酚作为防止甲基丙烯酸甲酯自聚所加的一定量的一种物质,它的定量检测很有必要。
背景:2,4-二甲基-6-叔丁基苯酚是一种能够快速与游离基反应并终止链反应的物质,通常被称为稳定剂。在储存或运输甲基丙烯酸甲酯时,添加适量的该物质可以防止甲基丙烯酸甲酯的自聚。若添加量过少,则无法有效阻止自聚;而添加量过多则会影响后续的聚合反应。因此,建立一种简便、快速、准确测定稳定剂含量的方法对于指导工艺运行具有非常重要的意义。
含量测定:
1. 方法一:
苏桂艳等人利用Rtx-wax毛细管柱,以乙腈为溶剂,氢火焰离子检测器检测,对工业用甲基丙烯酸甲酯中阻聚剂2,4-二甲基- 6-叔丁基苯酚含量进行测定。实验部分如下:
(1)色谱操作条件:
色谱柱:WCOT熔融石英15m×0.53 mm×1.0mm。进样口温度:250℃;检测器温度:250℃;柱温:初始温度100℃,保持 5min,以15℃/min速率加热到220℃,保持5min;分流比:1:10; 载气:氦气,恒压21.5kpa;进样量:1μL。
(2)校准曲线制备
标准储备溶液的配制:准确称取0.05g 2,4-二甲基-6-叔丁基苯酚置于50ml容量瓶中,用乙腈定容。
校验溶液的配制:分别称取0.1g,0.3g、0.5g标准储备溶液,精确至0.1mg,分别加入到50mL容量瓶,乙腈定容至刻度。
(3)试样分析步骤
试样混匀后直接注入色谱仪中测定。
实验结果为:该方法回收率为 98.5~101.5%,相对标准偏差为1.7~2.0%。此方法简便、快速、灵敏度高,并具有较好的准确度和精密度,适用于对甲基丙烯酸甲酯中阻聚剂2,4-二甲基-6-叔丁基苯酚的质控分析。
2. 方法二:
刘兴富等人提出用气相色谱外标法测定甲基丙烯酸甲酯中阻聚剂 2 ,4 -二甲基 - 6 -叔丁基苯酚的质量分数。实验部分如下:
(1)色谱条件:
色谱柱为SE-30填充柱,规格为2 m×3 mm;柱温130 ℃,汽化温度250 ℃;检测器温度250 ℃;柱前压40 kPa;灵敏度(RANGE)102;氢气流速60 mL/min;空气流速500 mL/min。
(2)工作曲线的绘制
精密称取0.010 7 g 2,4-二甲基-6-叔丁基苯酚标准品于100 mL容量瓶中,用甲醇稀释至刻度,摇匀。分别移取上述溶液2.5 mL、5 mL、10 mL、20 mL置于50 mL容量瓶中,用甲醇稀释至刻度,摇匀。待色谱仪稳定后进样,进样量均为2微升。
以峰面积对2,4-二甲基-6-叔丁基苯酚质量作图,绘制工作曲线。见图。
结果:采用气相色谱外标法测定甲基丙烯酸甲酯中阻聚剂2,4-二甲基-6-叔丁基苯酚的含量。在最佳底液条件下,加入一定量标准,对其在24 h内进行多次测定,定量结果的重复性好,且线性关系很好,说明此方法稳定性好。在甲基丙烯酸甲酯中分别加入2,4-二甲基-6-叔丁基苯酚标准,测得的平均回收率在96 %以上,说明该法用于甲基丙烯酸甲酯中的2,4-二甲基-6-叔丁基苯酚测定准确可靠。
参考文献:
[1]苏桂艳,王东川,苏建萍. 甲基丙烯酸甲酯中阻聚剂2,4-二甲基-6-叔丁基苯酚含量的测定 [J]. 化工管理, 2017, (23): 206.
[2]刘兴富,丛文俊,徐金贤. 甲基丙烯酸甲酯中阻聚剂2,4-二甲基-6-叔丁基苯酚的测定 [J]. 辽宁化工, 2004, (07): 432-433.
1
本文将讲述4-巯基苯硼酸在检测领域中有何应用,旨在为相关领域的研究人员提供参考思路。
简述:4-巯基苯硼酸是含巯基的有机硼酸类化合物,该类生物分子已经开始应用于硼中子捕获治疗(BNCT)领域。同时,有机硼酸类化合物被广泛应用于Suzuki反应中,用来构建各种C-C单键。
应用:
1. 检测唾液酸
微流控纸芯片技术由于其价廉、便捷、高效等众多优点,具有良好的应用前景,在食品安全检测、环境监测、疾病诊断等方面得到广泛应用。
张剑等人建立一种基于纸芯片的唾液酸检测体系。合成4-巯基苯硼酸修饰纳米金粒子(4-MPBA-AuNPS)。4-MPBA-AuNPS与唾液酸可发生选择性相互作用而逐渐团聚,颜色由红变蓝,在纸芯片上进行显色,手机拍照记录结果,Photoshop软件分析颜色强度。在0.9mmol·L-1~2.9mmol·L-1范围内,红蓝通道色度比值(ΔBlue/Red)与唾液酸浓度呈良好线性关系,检测限为1.9μmol·L-1,加样回收率为98.96%~101.1%。样品检测结果误差为-0.58%,准确度良好。该方法具有样品用量少、检测快速、准确性高等特点,有望用于人血清中唾液酸含量的检测。实验原理如下:
硼酸基通过可逆酯化反应与邻二醇类化合物进行选择性作用。可与SA分子中三元醇侧链发生反应,具有对SA进行特异性检测的潜力。研究利用4-巯基苯硼酸分子的巯基与AuNPs作用,将4-巯基苯硼酸修饰在纳米金颗粒表面(4-MPBA-AuNPS)制得SA识别探针,利用修饰纳米金表面的苯硼酸与SA之间的静电引力等相互作用,引起4-MPBA-AuNPS体系发生团聚,颜色由红变蓝,在纸芯片上进行显色。颜色变化速度与SA浓度成正比,根据红蓝通道色度比值(ΔBlue/Red)即可完成SA的快速检测。
2. 细菌定量检测
食品中的细菌超标会引发人体一系列反应,严重者会引起出血性腹泻、溶血性尿毒症和胆囊炎等肠道外感染和消化系统疾病。因此,开展细菌的快速检测分析对于保障食品卫生安全和人民群众身体健康具有重要意义。
王春鑫等人基于自主研发的智能手机比色分析系统,以4-巯基苯硼酸(4-Mercaptophenylboronic acid, 4-MPBA)修饰的金纳米粒子(4-MPBA-AuNPs)为检测探针,建立了一种高效、便捷、快速的细菌定量检测方法。4-MPBA-Au NPs中的硼酸基团可与细菌表面的多糖共价结合,从而达到识别细菌的目的。当4-MPBA-Au NPs溶液中含有的细菌浓度不同时,加入NaCl后,4-MPBA-AuNPs发生聚集的程度不同,其溶液的颜色也随之不同。因此,利用智能手机比色分析系统对4-MPBA-Au NPs溶液进行拍照和数字图片比色分析,可以实现细菌的定量检测。使用的4-MPBA-Au NPs纳米探针易制备、成本低且具有广谱性,可满足大批量样本的检测需求;智能手机比色分析系统的引入,有效减少了外界环境以及人为主观因素的干扰,提高了细菌比色分析的准确性;利用手机自定义App对图像RGB色度值进行数据分析,可以直接获得细菌的浓度,进一步提高细菌检测的时效性。本方法检测时间短(40 min),检测线性范围宽(104~109CFU/m L),检出限为5.74×102CFU/m L。猪肉样品中大肠杆菌O157∶H7的加标回收率为103.2%~115.7%,相对标准偏差为3.3%~7.3%。该方法成本低、易操作、易普及,为我国食品安全有效监管提供了一种新方法。
3. 检测亚硫酸根离子
高月滢等人通过可控自组装途径,开发一种基于4-巯基苯硼酸(4-mercaptoboric acid,4-MPBA)探针的表面增强拉曼散射(surface-enhanced Raman scattering,SERS)芯片的超灵敏亚硫酸根离子(SO32-)定量技术。该技术借助SO32-到SO42-的氧化转化特性,利用Lewis酸碱配位原理,建立基于特征峰1 382 cm-1与1 070 cm-1的比率型定量检测方程。该比率定量策略排除了SERS基底不均匀导致的检测误差,有效提高检测信号获取的可靠性,检出限低至5×10-8 mol/L,并在5×10-8~1×10-3 mol/L的浓度范围内呈良好线性关系(R2=0.997 4)。检测到白糖中SO32-含量为1.42 mg/kg,加标回收率在98.38%~107.82%之间,说明该方法对于实际样品中SO32-的痕量检测具有极大优势。
参考文献:
[1]张剑,许正雯,张博等. 4-巯基苯硼酸-纳米金粒子-纸芯片体系检测唾液酸 [J]. 化学研究与应用, 2023, 35 (05): 1238-1241.
[2]王春鑫,邓荣,牛晓峰等. 基于智能手机比色分析系统的细菌快速定量检测 [J]. 分析化学, 2023, 51 (08): 1302-1312. DOI:10.19756/j.issn.0253-3820.221516.
[3]高月滢,万玉琪,刘琳等. 表面增强拉曼光谱法 灵敏检测亚硫酸根离子 [J]. 食品科学, 2022, 43 (20): 328-335.
[4]大连双硼医药化工有限公司. 一种合成4-巯基苯硼酸的工艺方法. 2022-02-18.
显示全部本文将讲述4-巯基苯硼酸在检测领域中有何应用,旨在为相关领域的研究人员提供参考思路。
简述:4-巯基苯硼酸是含巯基的有机硼酸类化合物,该类生物分子已经开始应用于硼中子捕获治疗(BNCT)领域。同时,有机硼酸类化合物被广泛应用于Suzuki反应中,用来构建各种C-C单键。
应用:
1. 检测唾液酸
微流控纸芯片技术由于其价廉、便捷、高效等众多优点,具有良好的应用前景,在食品安全检测、环境监测、疾病诊断等方面得到广泛应用。
张剑等人建立一种基于纸芯片的唾液酸检测体系。合成4-巯基苯硼酸修饰纳米金粒子(4-MPBA-AuNPS)。4-MPBA-AuNPS与唾液酸可发生选择性相互作用而逐渐团聚,颜色由红变蓝,在纸芯片上进行显色,手机拍照记录结果,Photoshop软件分析颜色强度。在0.9mmol·L-1~2.9mmol·L-1范围内,红蓝通道色度比值(ΔBlue/Red)与唾液酸浓度呈良好线性关系,检测限为1.9μmol·L-1,加样回收率为98.96%~101.1%。样品检测结果误差为-0.58%,准确度良好。该方法具有样品用量少、检测快速、准确性高等特点,有望用于人血清中唾液酸含量的检测。实验原理如下:
硼酸基通过可逆酯化反应与邻二醇类化合物进行选择性作用。可与SA分子中三元醇侧链发生反应,具有对SA进行特异性检测的潜力。研究利用4-巯基苯硼酸分子的巯基与AuNPs作用,将4-巯基苯硼酸修饰在纳米金颗粒表面(4-MPBA-AuNPS)制得SA识别探针,利用修饰纳米金表面的苯硼酸与SA之间的静电引力等相互作用,引起4-MPBA-AuNPS体系发生团聚,颜色由红变蓝,在纸芯片上进行显色。颜色变化速度与SA浓度成正比,根据红蓝通道色度比值(ΔBlue/Red)即可完成SA的快速检测。
2. 细菌定量检测
食品中的细菌超标会引发人体一系列反应,严重者会引起出血性腹泻、溶血性尿毒症和胆囊炎等肠道外感染和消化系统疾病。因此,开展细菌的快速检测分析对于保障食品卫生安全和人民群众身体健康具有重要意义。
王春鑫等人基于自主研发的智能手机比色分析系统,以4-巯基苯硼酸(4-Mercaptophenylboronic acid, 4-MPBA)修饰的金纳米粒子(4-MPBA-AuNPs)为检测探针,建立了一种高效、便捷、快速的细菌定量检测方法。4-MPBA-Au NPs中的硼酸基团可与细菌表面的多糖共价结合,从而达到识别细菌的目的。当4-MPBA-Au NPs溶液中含有的细菌浓度不同时,加入NaCl后,4-MPBA-AuNPs发生聚集的程度不同,其溶液的颜色也随之不同。因此,利用智能手机比色分析系统对4-MPBA-Au NPs溶液进行拍照和数字图片比色分析,可以实现细菌的定量检测。使用的4-MPBA-Au NPs纳米探针易制备、成本低且具有广谱性,可满足大批量样本的检测需求;智能手机比色分析系统的引入,有效减少了外界环境以及人为主观因素的干扰,提高了细菌比色分析的准确性;利用手机自定义App对图像RGB色度值进行数据分析,可以直接获得细菌的浓度,进一步提高细菌检测的时效性。本方法检测时间短(40 min),检测线性范围宽(104~109CFU/m L),检出限为5.74×102CFU/m L。猪肉样品中大肠杆菌O157∶H7的加标回收率为103.2%~115.7%,相对标准偏差为3.3%~7.3%。该方法成本低、易操作、易普及,为我国食品安全有效监管提供了一种新方法。
3. 检测亚硫酸根离子
高月滢等人通过可控自组装途径,开发一种基于4-巯基苯硼酸(4-mercaptoboric acid,4-MPBA)探针的表面增强拉曼散射(surface-enhanced Raman scattering,SERS)芯片的超灵敏亚硫酸根离子(SO32-)定量技术。该技术借助SO32-到SO42-的氧化转化特性,利用Lewis酸碱配位原理,建立基于特征峰1 382 cm-1与1 070 cm-1的比率型定量检测方程。该比率定量策略排除了SERS基底不均匀导致的检测误差,有效提高检测信号获取的可靠性,检出限低至5×10-8 mol/L,并在5×10-8~1×10-3 mol/L的浓度范围内呈良好线性关系(R2=0.997 4)。检测到白糖中SO32-含量为1.42 mg/kg,加标回收率在98.38%~107.82%之间,说明该方法对于实际样品中SO32-的痕量检测具有极大优势。
参考文献:
[1]张剑,许正雯,张博等. 4-巯基苯硼酸-纳米金粒子-纸芯片体系检测唾液酸 [J]. 化学研究与应用, 2023, 35 (05): 1238-1241.
[2]王春鑫,邓荣,牛晓峰等. 基于智能手机比色分析系统的细菌快速定量检测 [J]. 分析化学, 2023, 51 (08): 1302-1312. DOI:10.19756/j.issn.0253-3820.221516.
[3]高月滢,万玉琪,刘琳等. 表面增强拉曼光谱法 灵敏检测亚硫酸根离子 [J]. 食品科学, 2022, 43 (20): 328-335.
[4]大连双硼医药化工有限公司. 一种合成4-巯基苯硼酸的工艺方法. 2022-02-18.
1
准确检测白芥子中对羟基苯乙腈的含量对于质量控制和药物疗效评估至关重要。
背景:对羟基苯乙腈广泛存在于中药炒白芥子中,是一种重要的医药中间体。白芥子为2010版《中国药典》收载中药,为十字花科(Cruciferae)芥属植物白芥(Sinapis alba L.)的干燥成熟种子,辛,温,归肺经,具有温肺豁痰利气、 结、通络、止痛等功效。
关于白芥子中对羟基苯乙腈含量的变化
1. 炒制前后白芥子中对羟基苯乙腈含量的变化
杨雪萍等人对白芥子按照2005年版《中华人民共和国药典》规定的清炒法进行炒制,得到5 min和10 min的炒制品,然后对生、炒白芥子用石油醚脱脂,再用乙醇提取。采用RP-HPLC测定白芥子生品和炒制品中对羟基苯乙腈的含量。实验方法为:
(1)炒制品的制备
取净白芥子,置炒制容器内,用文火加热,炒至表面呈黄色,有爆裂声,并放出香辣气味,取出,放凉。作者分别制备了5min和10min炒制品。取生白芥子与炒制品各20g,粉碎,置具塞瓶中,加入石油醚脱脂,过滤,药渣用体积分数为95%乙醇回流提取3次,合并提取液;浓缩至无醇味,得其浸膏。
(2)对照品溶液的制备
精密称取对羟基苯乙腈对照品,加甲醇溶解,并定容至50 mL容量瓶中,得质量浓度为5.0g·L-1的溶液,备用。
(3)色谱条件
色谱柱为Luna 5uPheny1-Hexy1(250mm×4.60mm,5μm),流动相为甲醇-水(体积比为35∶65)流速为0.6 mL·min-1,检测波长为287nm,柱温为室温。对照品和样品的色谱图见图。
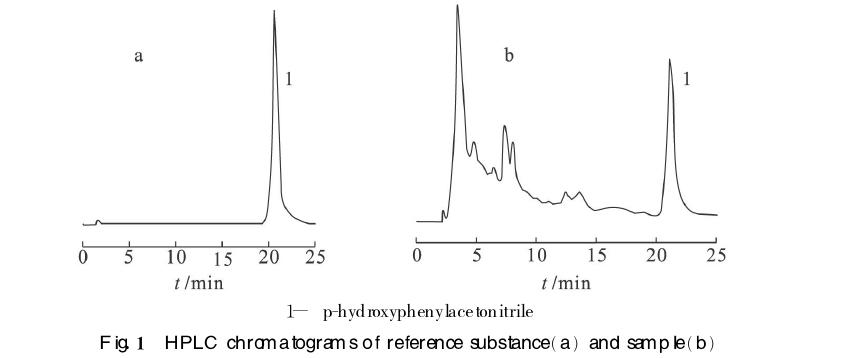
结果为白芥子生品所得浸膏中对羟基苯乙腈的含量质量分数是0.76%,5 min白芥子炒制品所得浸膏中对羟基苯乙腈的含量质量分数为16.53%,10 min白芥子炒制品所得浸膏中对羟基苯乙腈的含量质量分数是21.44%。结果表明炒制可导致白芥子中对羟基苯乙腈的含量显著增加。
2. 不同炮制时间对白芥子中对羟基苯乙腈含量的变化影响
李怡萱等人建立了超高效液相色谱法(UPLC)同时测定炒白芥子中有效成分(羟基苯乙腈和芥子碱硫氰酸盐)含量的方法,探究不同炮制时间对白芥子中活性成分含量的影响。方法:采用UPLC法与色谱柱为Waters BEH C1 8 色 谱柱(2.1 mm×100 mm,1.7μm),以乙腈-0.02 mol/L磷酸二氢钾溶液为流动相梯度洗脱,检测波长为222 nm,流速为0.3 mL/min,柱温为25℃,进样量为2μL,测定其白芥子中对羟基苯乙腈和芥子碱硫氰酸盐的含量。色谱条件和溶液配制具体如下:
2.1 色谱条件
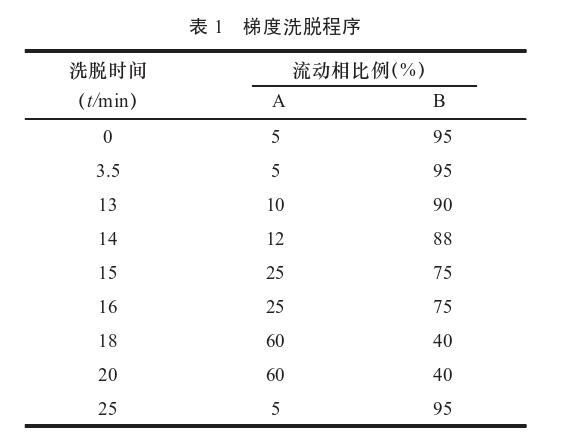
色谱柱为Waters BEH C18 色谱柱(2.1 mm× 100 mm,1.7μm);流动相为乙腈(A)-0.02 mol/L 磷酸二氢钾水溶液(B),依照表1进行梯度洗脱, 并在检测波长222 nm,流速为0.3 mL/min,柱温为25℃,室温为15℃下,进样量2μL,按WatersAcquity H-class型超高效液相色谱仪说明书中方法 测定其含量。详见表1。
2.2 溶液的制备
(1)对照品溶液的制备
分别取对羟基苯乙腈、芥子碱硫氰酸盐对照品适量,精密称定,置于20 mL 容量瓶中,加50%甲醇液溶解,定容至刻度,即得各成分单一对照品储备液,备用。精密移取上述对照品储备液各1 mL,置于同 一5 mL容量瓶中,加50%甲醇液稀释,定容至刻度,即得对羟基苯乙腈浓度63.07μg/mL和芥子碱硫氰酸盐147.18μg/mL的混合对照品溶液。
(2)供试品溶液的制备
取炒白芥子样品粉末(过4号筛)0.3 g,精密称定,置于具塞锥形瓶中,加 50%甲醇液25 mL,称定其质量,超声提取30 min,取出放冷,再次称其质量并用50%甲醇液补足其原有质量,摇匀,经0.22μm微孔滤膜滤过,取续滤液,即得。
(3)空白对照溶液的制备
以50%甲醇液为空白对照溶液。
结果:羟基苯乙腈和芥子碱硫氰酸盐浓度分别在0.006 307~0.1 577μg/mL(r2=1,n=6)、0.01 472~0.3 680μg/mL(r2=1,n=6)范围内呈良好的线性关系。该炒白芥子的含量测定方法,操作简单,结果准确,具有较好的可重复性和稳定性,为炒白芥子饮片的质量控制提供依据。
参考文献:
[1]李怡萱,盛一梁,吴嫣等. 基于超高效液相色谱定量测定法探究不同炮制时间对白芥子中有效成分含量的影响 [J]. 抗感染药学, 2021, 18 (03): 316-320. DOI:10.13493/j.issn.1672-7878.2021.03-003.
[2]冯宝民,邱琳,谌启鹏等. 基于炮效关系研究白芥子镇咳药效物质基础 [J]. 中国药理学通报, 2010, 26 (09): 1173-1176.
[3]杨雪萍,王惠国,冯宝民等. 白芥子炒制前后对羟基苯乙腈含量的变化 [J]. 沈阳药科大学学报, 2010, 27 (08): 623-625. DOI:10.14066/j.cnki.cn21-1349/r.2010.08.009.
显示全部准确检测白芥子中对羟基苯乙腈的含量对于质量控制和药物疗效评估至关重要。
背景:对羟基苯乙腈广泛存在于中药炒白芥子中,是一种重要的医药中间体。白芥子为2010版《中国药典》收载中药,为十字花科(Cruciferae)芥属植物白芥(Sinapis alba L.)的干燥成熟种子,辛,温,归肺经,具有温肺豁痰利气、 结、通络、止痛等功效。
关于白芥子中对羟基苯乙腈含量的变化
1. 炒制前后白芥子中对羟基苯乙腈含量的变化
杨雪萍等人对白芥子按照2005年版《中华人民共和国药典》规定的清炒法进行炒制,得到5 min和10 min的炒制品,然后对生、炒白芥子用石油醚脱脂,再用乙醇提取。采用RP-HPLC测定白芥子生品和炒制品中对羟基苯乙腈的含量。实验方法为:
(1)炒制品的制备
取净白芥子,置炒制容器内,用文火加热,炒至表面呈黄色,有爆裂声,并放出香辣气味,取出,放凉。作者分别制备了5min和10min炒制品。取生白芥子与炒制品各20g,粉碎,置具塞瓶中,加入石油醚脱脂,过滤,药渣用体积分数为95%乙醇回流提取3次,合并提取液;浓缩至无醇味,得其浸膏。
(2)对照品溶液的制备
精密称取对羟基苯乙腈对照品,加甲醇溶解,并定容至50 mL容量瓶中,得质量浓度为5.0g·L-1的溶液,备用。
(3)色谱条件
色谱柱为Luna 5uPheny1-Hexy1(250mm×4.60mm,5μm),流动相为甲醇-水(体积比为35∶65)流速为0.6 mL·min-1,检测波长为287nm,柱温为室温。对照品和样品的色谱图见图。
结果为白芥子生品所得浸膏中对羟基苯乙腈的含量质量分数是0.76%,5 min白芥子炒制品所得浸膏中对羟基苯乙腈的含量质量分数为16.53%,10 min白芥子炒制品所得浸膏中对羟基苯乙腈的含量质量分数是21.44%。结果表明炒制可导致白芥子中对羟基苯乙腈的含量显著增加。
2. 不同炮制时间对白芥子中对羟基苯乙腈含量的变化影响
李怡萱等人建立了超高效液相色谱法(UPLC)同时测定炒白芥子中有效成分(羟基苯乙腈和芥子碱硫氰酸盐)含量的方法,探究不同炮制时间对白芥子中活性成分含量的影响。方法:采用UPLC法与色谱柱为Waters BEH C1 8 色 谱柱(2.1 mm×100 mm,1.7μm),以乙腈-0.02 mol/L磷酸二氢钾溶液为流动相梯度洗脱,检测波长为222 nm,流速为0.3 mL/min,柱温为25℃,进样量为2μL,测定其白芥子中对羟基苯乙腈和芥子碱硫氰酸盐的含量。色谱条件和溶液配制具体如下:
2.1 色谱条件
色谱柱为Waters BEH C18 色谱柱(2.1 mm× 100 mm,1.7μm);流动相为乙腈(A)-0.02 mol/L 磷酸二氢钾水溶液(B),依照表1进行梯度洗脱, 并在检测波长222 nm,流速为0.3 mL/min,柱温为25℃,室温为15℃下,进样量2μL,按WatersAcquity H-class型超高效液相色谱仪说明书中方法 测定其含量。详见表1。
2.2 溶液的制备
(1)对照品溶液的制备
分别取对羟基苯乙腈、芥子碱硫氰酸盐对照品适量,精密称定,置于20 mL 容量瓶中,加50%甲醇液溶解,定容至刻度,即得各成分单一对照品储备液,备用。精密移取上述对照品储备液各1 mL,置于同 一5 mL容量瓶中,加50%甲醇液稀释,定容至刻度,即得对羟基苯乙腈浓度63.07μg/mL和芥子碱硫氰酸盐147.18μg/mL的混合对照品溶液。
(2)供试品溶液的制备
取炒白芥子样品粉末(过4号筛)0.3 g,精密称定,置于具塞锥形瓶中,加 50%甲醇液25 mL,称定其质量,超声提取30 min,取出放冷,再次称其质量并用50%甲醇液补足其原有质量,摇匀,经0.22μm微孔滤膜滤过,取续滤液,即得。
(3)空白对照溶液的制备
以50%甲醇液为空白对照溶液。
结果:羟基苯乙腈和芥子碱硫氰酸盐浓度分别在0.006 307~0.1 577μg/mL(r2=1,n=6)、0.01 472~0.3 680μg/mL(r2=1,n=6)范围内呈良好的线性关系。该炒白芥子的含量测定方法,操作简单,结果准确,具有较好的可重复性和稳定性,为炒白芥子饮片的质量控制提供依据。
参考文献:
[1]李怡萱,盛一梁,吴嫣等. 基于超高效液相色谱定量测定法探究不同炮制时间对白芥子中有效成分含量的影响 [J]. 抗感染药学, 2021, 18 (03): 316-320. DOI:10.13493/j.issn.1672-7878.2021.03-003.
[2]冯宝民,邱琳,谌启鹏等. 基于炮效关系研究白芥子镇咳药效物质基础 [J]. 中国药理学通报, 2010, 26 (09): 1173-1176.
[3]杨雪萍,王惠国,冯宝民等. 白芥子炒制前后对羟基苯乙腈含量的变化 [J]. 沈阳药科大学学报, 2010, 27 (08): 623-625. DOI:10.14066/j.cnki.cn21-1349/r.2010.08.009.
1
本文将介绍杆菌肽含量测定的方法,以便准确、可靠地确定杆菌肽的含量。
简述:杆菌肽(Bacitracin)是一类由非核糖体肽合成酶(Nonribosomal peptide synthetase, NRPS)催化合成的环状多肽抗生素。地衣芽孢杆菌(Bacillus licheniformis)是杆菌肽的主要生产宿主。
随着科学的不断进步,人们对杆菌肽特性的了解也越来越深入,杆菌肽的应用必将会随着研究的不断深入而更加广泛。国外杆菌肽定量测定方法常采用微生物检定法,如管碟法、浊度法以及高效液相色谱定量测定法,且这些方法均已被国际药典和各国药典收录。国内常用的方法参考药典,有管碟法、浊度法等。
含量测定:
1. HPLC结合组分制备对杆菌肽中有关物质及有效组分含量测定
张含智等人建立基于HPLC结合组分制备对杆菌肽中有关物质及有效组分含量测定的分析方法。方法为:采用Agilent HPLC,选择Diamonsil Plus C1 8 色谱柱(4.6 mm×250 mm,5μm),以50 mmol·L-1 甲酸铵溶液(p H经甲酸调至4.0)为流动相 A,乙腈为流动相B,流速为1 m L·min-1 ,线性梯度洗脱,检测波长为254 nm。HPLC条件与溶液配制具体为:
(1 )HPLC条件
按《美国药典》40版中杆菌肽的有关物质的分析方法,选择Diamonsil Plus C1 8 色谱柱(4.6 mm× 250 mm,5μm),以磷酸氢二钾、磷酸二氢钾-甲醇- 乙腈为流动相,流速为1 m L·min-1 ;柱温为30℃; 进样量为20μL;检测波长为254 nm。在组分制备过程中,进样量调整为100μL。
实验中以50 mmol·L-1 甲酸铵溶液(用甲酸调节pH值至4.0)为流动相A,乙腈为流动相B,按表2线性梯度洗脱,流速为1 m L·min-1 ;柱温为30 ℃;进样量为20μL;检测波长为254 nm。

(2)溶液配制
精密称取杆菌肽锌对照品及杆菌肽锌供试品 20 mg,置10 mL量瓶中,加乙二胺四乙酸二钠(ED TA-2Na)溶液(40 g·L-1,pH经氢氧化钠溶液调至 7.0)溶解并定量稀释至刻度,摇匀。杆菌肽原料药供试品用水-乙腈(77∶23)溶解,其他同于杆菌肽锌。杆菌肽原料药制备溶液质量浓度为15 mg·m L-1 ,进样量为100μL。
该液相分析方法可同时测定杆菌肽中的有关物质及有效组分含量,适用于该产品的质量控制。
2. 基于比浊法的杆菌肽定量测定
杆菌肽在研究应用过程中,定量测定方法不统一,结果缺乏参考性。为规范其测定方法,王志新等人通过建立杆菌肽浓度,对数值与OD6 00之间的线性关系,以重复性和精密度为指标,优化指示菌初始浓度、杆菌肽溶液与菌悬液的比例、培养时间等因素,确定比浊法测定杆菌肽抑菌活性的方法。结果显示,比浊法的最适测定条件为:指示菌初始浓度107 CFU/mL,杆菌肽溶液与菌悬液比例1∶9,培养时间4 h,在此条件下,线性关系良好,R2达到0.99以上,且具有良好的重复性。进一步选用大肠杆菌ATCC 44752和金黄色葡萄球菌ATCC 25923进行研究,结果重复性好,精密度高。研究结果将为比浊法的进一步应用以及杆菌肽在试验和生产过程中的定量测定提供参考和依据。
参考文献:
[1]吴志勇. 杆菌肽A合成机制及其NRPS功能结构域研究[D]. 江南大学, 2022. DOI:10.27169/d.cnki.gwqgu.2022.002312.
[2]王志新,刘洋,周景波等. 基于比浊法的杆菌肽定量测定方法优化 [J]. 生物技术通报, 2020, 36 (05): 92-97. DOI:10.13560/j.cnki.biotech.bull.1985.2019-0994.
[3]张含智,秦峰,刘浩. HPLC结合组分制备分析杆菌肽中的有关物质及有效组分含量 [J]. 中国药学杂志, 2018, 53 (23): 2041-2046.
显示全部本文将介绍杆菌肽含量测定的方法,以便准确、可靠地确定杆菌肽的含量。
简述:杆菌肽(Bacitracin)是一类由非核糖体肽合成酶(Nonribosomal peptide synthetase, NRPS)催化合成的环状多肽抗生素。地衣芽孢杆菌(Bacillus licheniformis)是杆菌肽的主要生产宿主。
随着科学的不断进步,人们对杆菌肽特性的了解也越来越深入,杆菌肽的应用必将会随着研究的不断深入而更加广泛。国外杆菌肽定量测定方法常采用微生物检定法,如管碟法、浊度法以及高效液相色谱定量测定法,且这些方法均已被国际药典和各国药典收录。国内常用的方法参考药典,有管碟法、浊度法等。
含量测定:
1. HPLC结合组分制备对杆菌肽中有关物质及有效组分含量测定
张含智等人建立基于HPLC结合组分制备对杆菌肽中有关物质及有效组分含量测定的分析方法。方法为:采用Agilent HPLC,选择Diamonsil Plus C1 8 色谱柱(4.6 mm×250 mm,5μm),以50 mmol·L-1 甲酸铵溶液(p H经甲酸调至4.0)为流动相 A,乙腈为流动相B,流速为1 m L·min-1 ,线性梯度洗脱,检测波长为254 nm。HPLC条件与溶液配制具体为:
(1 )HPLC条件
按《美国药典》40版中杆菌肽的有关物质的分析方法,选择Diamonsil Plus C1 8 色谱柱(4.6 mm× 250 mm,5μm),以磷酸氢二钾、磷酸二氢钾-甲醇- 乙腈为流动相,流速为1 m L·min-1 ;柱温为30℃; 进样量为20μL;检测波长为254 nm。在组分制备过程中,进样量调整为100μL。
实验中以50 mmol·L-1 甲酸铵溶液(用甲酸调节pH值至4.0)为流动相A,乙腈为流动相B,按表2线性梯度洗脱,流速为1 m L·min-1 ;柱温为30 ℃;进样量为20μL;检测波长为254 nm。
(2)溶液配制
精密称取杆菌肽锌对照品及杆菌肽锌供试品 20 mg,置10 mL量瓶中,加乙二胺四乙酸二钠(ED TA-2Na)溶液(40 g·L-1,pH经氢氧化钠溶液调至 7.0)溶解并定量稀释至刻度,摇匀。杆菌肽原料药供试品用水-乙腈(77∶23)溶解,其他同于杆菌肽锌。杆菌肽原料药制备溶液质量浓度为15 mg·m L-1 ,进样量为100μL。
该液相分析方法可同时测定杆菌肽中的有关物质及有效组分含量,适用于该产品的质量控制。
2. 基于比浊法的杆菌肽定量测定
杆菌肽在研究应用过程中,定量测定方法不统一,结果缺乏参考性。为规范其测定方法,王志新等人通过建立杆菌肽浓度,对数值与OD6 00之间的线性关系,以重复性和精密度为指标,优化指示菌初始浓度、杆菌肽溶液与菌悬液的比例、培养时间等因素,确定比浊法测定杆菌肽抑菌活性的方法。结果显示,比浊法的最适测定条件为:指示菌初始浓度107 CFU/mL,杆菌肽溶液与菌悬液比例1∶9,培养时间4 h,在此条件下,线性关系良好,R2达到0.99以上,且具有良好的重复性。进一步选用大肠杆菌ATCC 44752和金黄色葡萄球菌ATCC 25923进行研究,结果重复性好,精密度高。研究结果将为比浊法的进一步应用以及杆菌肽在试验和生产过程中的定量测定提供参考和依据。
参考文献:
[1]吴志勇. 杆菌肽A合成机制及其NRPS功能结构域研究[D]. 江南大学, 2022. DOI:10.27169/d.cnki.gwqgu.2022.002312.
[2]王志新,刘洋,周景波等. 基于比浊法的杆菌肽定量测定方法优化 [J]. 生物技术通报, 2020, 36 (05): 92-97. DOI:10.13560/j.cnki.biotech.bull.1985.2019-0994.
[3]张含智,秦峰,刘浩. HPLC结合组分制备分析杆菌肽中的有关物质及有效组分含量 [J]. 中国药学杂志, 2018, 53 (23): 2041-2046.
1
本文将讨论如何用2,6-二氯靛酚钠法进行维生素C的定量测定,以期为相关领域的研究人员提供实验思路。
简述:2,6-二氯靛酚钠,英文名称:2,6-Dichloroindophenol sodium salt,CAS:620-45-1,分子式:C12H6Cl2NNaO2,外观与性状:无气味的暗绿色固体。2,6-二氯靛酚钠常用于抗坏血酸的测定,还用作氧化还原指示剂和色谱分析试剂。
应用:
1. 测定青椒中抗坏血酸
维生素C也称抗坏血酸,是一种水溶性有机化合物,虽然没有游离羟基,但是其在碱性环境下很容易被氧化,故具备有机酸的一些性。在追求品质的当代,人们对于微量元素的有效补充越来越注重。食品中抗坏血酸的测定方法有很多,国标GB 5009.86—2016中第一法为高效液相色谱法,第二法为荧光法,第三法为2,6-二氯靛酚法等等。其中,2,6-二氯靛酚法具有不需要大型分析仪器设备、操作简单、试剂价格便宜、精密度和准确度高等优点,广泛用于水果、蔬菜及其制品中L-抗坏血酸的测定。
刘晶等人研究2,6-二氯靛酚法测定青椒中抗坏血酸的脱色条件,主要对比了活性炭、皂土和高岭土3种吸附剂对青椒中抗坏血酸吸附率及对叶绿素的脱除率的影响。试验方法如下:
(1)抗坏血酸吸附试验
取50 m L 0.2 mg/m L的抗坏血酸溶液于100 mL 烧杯中,加入一定质量的皂土、活性炭和高岭土,将烧杯分别置于一定温度下的磁力搅拌器中恒温搅拌,过滤,采用2,6-二氯靛酚滴定法测定滤液中抗坏血酸的含量。
(2)叶绿素吸附试验
取5 g新鲜的青椒,加入少许提取剂(无水乙醇∶丙酮=1∶1)进行研磨后定容至100 m L,然后将其转移至烧杯中,加入一定质量的皂土、活性炭和高岭土,将烧杯分别置于一定温度下的磁力搅拌器中恒温搅拌,过滤,参照NY/T 3082—2017中的方法测定溶液中的叶绿素含量。
结果表明,以4%的皂土为吸附剂,在25℃下吸附25 min,对抗坏血酸的吸附率为5.1%,对青椒叶绿素的脱除率达86.1%,经脱色处理后的样品基本上无颜色,且抗坏血酸的吸附量小。采用2,6-二氯靛酚法测定其中的抗坏血酸含量,终点颜色易于判断且基本不影响分析结果,该方法精密度为 4.17%,回收率在97.7%~107.0%,能够满足测定要求。
2. 测定鲜榨桃汁中维生素C的含量
通过2,6-二氯靛酚滴定法测定鲜榨桃汁中维生素C的含量,可以有效判定鲜榨桃汁品质的优劣。
张彩珍通过常规处理完成鲜榨桃汁的制作,在避光条件下,运用2,6-二氯靛酚滴定法测定鲜榨桃汁中维生素C含量,并对测定精密度进行检验。经测定,鲜榨桃汁中维生素C含量为5.38 mg/100 g,3组平行样精密度RSD=0.10%。该方法相对于其他方法来说, 具有结果可视性强、前处理步骤简单等优势,且测定精密度RSD≤0.5%,满足相关要求,可以作为检测鲜榨桃汁维生素C的分析方法。
3. 测定蜂胶中维生素C
陈光等人用2,6-二氯靛酚滴定法测定蜂胶中维生素C,试验方法如下:称取待测样品0.500 0g置于100mL容量瓶中, 加入无水乙醇30mL, 置于HY-5型回旋振荡器上, 以50r·min-1速率振荡至样品完全溶解, 取下用乙二酸溶液稀释至刻度, 摇匀待测。移取待测液10.00mL置于50 mL白色陶瓷蒸发皿中, 用已标定过的2, 6-二氯靛酚标准溶液滴定, 边滴定边用玻璃棒搅拌, 直至溶液呈粉红色保留15s不褪色即为终点。同时做空白试验。维生素C的质量分数按式 (1) 计算:
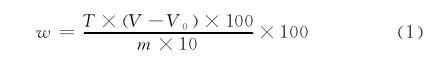
式中:w为维生素C质量分数, %;T为2, 6-二氯靛酚溶液的滴定度, g·mL-1;V为滴定样品溶液所消耗2, 6-二氯靛酚标准溶液的体积, mL;V0为滴定空白溶液所消耗2, 6-二氯靛酚溶液标准的体积, mL;m为称样量, g。
该方法用无水乙醇溶解蜂胶样品, 用乙二酸提取和固定维生素C, 并且使样品溶液呈淡黄色, 同时用白色陶瓷蒸发皿代替三角瓶滴定, 有助于判断滴定终点, 方法操作简便、快速、可用于蜂胶及其产品中维生素C的含量的测定。
参考文献:
[1]刘晶,李会. 2,6-二氯靛酚法测定青椒中抗坏血酸的脱色条件研究 [J]. 农产品加工, 2022, (07): 72-74. DOI:10.16693/j.cnki.1671-9646(X).2022.04.014.
[2]张彩珍. 2,6-二氯靛酚滴定法测定鲜榨桃汁中维生素C含量 [J]. 食品安全导刊, 2021, (24): 53+55. DOI:10.16043/j.cnki.cfs.2021.24.033.
[3]陈光,孙妍. 2,6-二氯靛酚滴定法测定蜂胶中维生素C [J]. 理化检验(化学分册), 2014, 50 (08): 1041-1042.
显示全部本文将讨论如何用2,6-二氯靛酚钠法进行维生素C的定量测定,以期为相关领域的研究人员提供实验思路。
简述:2,6-二氯靛酚钠,英文名称:2,6-Dichloroindophenol sodium salt,CAS:620-45-1,分子式:C12H6Cl2NNaO2,外观与性状:无气味的暗绿色固体。2,6-二氯靛酚钠常用于抗坏血酸的测定,还用作氧化还原指示剂和色谱分析试剂。
应用:
1. 测定青椒中抗坏血酸
维生素C也称抗坏血酸,是一种水溶性有机化合物,虽然没有游离羟基,但是其在碱性环境下很容易被氧化,故具备有机酸的一些性。在追求品质的当代,人们对于微量元素的有效补充越来越注重。食品中抗坏血酸的测定方法有很多,国标GB 5009.86—2016中第一法为高效液相色谱法,第二法为荧光法,第三法为2,6-二氯靛酚法等等。其中,2,6-二氯靛酚法具有不需要大型分析仪器设备、操作简单、试剂价格便宜、精密度和准确度高等优点,广泛用于水果、蔬菜及其制品中L-抗坏血酸的测定。
刘晶等人研究2,6-二氯靛酚法测定青椒中抗坏血酸的脱色条件,主要对比了活性炭、皂土和高岭土3种吸附剂对青椒中抗坏血酸吸附率及对叶绿素的脱除率的影响。试验方法如下:
(1)抗坏血酸吸附试验
取50 m L 0.2 mg/m L的抗坏血酸溶液于100 mL 烧杯中,加入一定质量的皂土、活性炭和高岭土,将烧杯分别置于一定温度下的磁力搅拌器中恒温搅拌,过滤,采用2,6-二氯靛酚滴定法测定滤液中抗坏血酸的含量。
(2)叶绿素吸附试验
取5 g新鲜的青椒,加入少许提取剂(无水乙醇∶丙酮=1∶1)进行研磨后定容至100 m L,然后将其转移至烧杯中,加入一定质量的皂土、活性炭和高岭土,将烧杯分别置于一定温度下的磁力搅拌器中恒温搅拌,过滤,参照NY/T 3082—2017中的方法测定溶液中的叶绿素含量。
结果表明,以4%的皂土为吸附剂,在25℃下吸附25 min,对抗坏血酸的吸附率为5.1%,对青椒叶绿素的脱除率达86.1%,经脱色处理后的样品基本上无颜色,且抗坏血酸的吸附量小。采用2,6-二氯靛酚法测定其中的抗坏血酸含量,终点颜色易于判断且基本不影响分析结果,该方法精密度为 4.17%,回收率在97.7%~107.0%,能够满足测定要求。
2. 测定鲜榨桃汁中维生素C的含量
通过2,6-二氯靛酚滴定法测定鲜榨桃汁中维生素C的含量,可以有效判定鲜榨桃汁品质的优劣。
张彩珍通过常规处理完成鲜榨桃汁的制作,在避光条件下,运用2,6-二氯靛酚滴定法测定鲜榨桃汁中维生素C含量,并对测定精密度进行检验。经测定,鲜榨桃汁中维生素C含量为5.38 mg/100 g,3组平行样精密度RSD=0.10%。该方法相对于其他方法来说, 具有结果可视性强、前处理步骤简单等优势,且测定精密度RSD≤0.5%,满足相关要求,可以作为检测鲜榨桃汁维生素C的分析方法。
3. 测定蜂胶中维生素C
陈光等人用2,6-二氯靛酚滴定法测定蜂胶中维生素C,试验方法如下:称取待测样品0.500 0g置于100mL容量瓶中, 加入无水乙醇30mL, 置于HY-5型回旋振荡器上, 以50r·min-1速率振荡至样品完全溶解, 取下用乙二酸溶液稀释至刻度, 摇匀待测。移取待测液10.00mL置于50 mL白色陶瓷蒸发皿中, 用已标定过的2, 6-二氯靛酚标准溶液滴定, 边滴定边用玻璃棒搅拌, 直至溶液呈粉红色保留15s不褪色即为终点。同时做空白试验。维生素C的质量分数按式 (1) 计算:
式中:w为维生素C质量分数, %;T为2, 6-二氯靛酚溶液的滴定度, g·mL-1;V为滴定样品溶液所消耗2, 6-二氯靛酚标准溶液的体积, mL;V0为滴定空白溶液所消耗2, 6-二氯靛酚溶液标准的体积, mL;m为称样量, g。
该方法用无水乙醇溶解蜂胶样品, 用乙二酸提取和固定维生素C, 并且使样品溶液呈淡黄色, 同时用白色陶瓷蒸发皿代替三角瓶滴定, 有助于判断滴定终点, 方法操作简便、快速、可用于蜂胶及其产品中维生素C的含量的测定。
参考文献:
[1]刘晶,李会. 2,6-二氯靛酚法测定青椒中抗坏血酸的脱色条件研究 [J]. 农产品加工, 2022, (07): 72-74. DOI:10.16693/j.cnki.1671-9646(X).2022.04.014.
[2]张彩珍. 2,6-二氯靛酚滴定法测定鲜榨桃汁中维生素C含量 [J]. 食品安全导刊, 2021, (24): 53+55. DOI:10.16043/j.cnki.cfs.2021.24.033.
[3]陈光,孙妍. 2,6-二氯靛酚滴定法测定蜂胶中维生素C [J]. 理化检验(化学分册), 2014, 50 (08): 1041-1042.
1
了解1-苯基-1,2-丙二酮关于振动光谱和理论计算的相关研究,对于正确使用和认识其潜在风险非常重要。
背景:1-苯基-1,2-丙二酮(1-phenyl-1,2-propanedione,PPD)是一种典型的α-二羰基化合物,除了用于不对称有机合成反应外,还可用作光致聚合牙齿树脂复合材料的光敏剂。Leonard等人研究了PPD分子n-π*的电子吸收光谱,发现扭转角度对PPD的吸收有很大影响。SusyLopes等人研究了低温基质隔离下PPD的分子异构化以及紫外诱发的PPD去羰基化现象,进一步探讨了PPD的光化学和光物理现象。
振动光谱和理论计算研究:
利用共振拉曼光谱技术结合密度泛函理论计算,研究了PPD的激发态结构动力学,通过分析振动光谱和电子吸收光谱,考察PPD在激发态时分子结构以及电子振动耦合吸收等方面的信息。
(1)实验方法:
分别以环己烷和乙腈做溶剂,配制PPD溶液的浓度约为3.00~4.00 mmol/L。通过四倍频激光线 (波长266nm) 和氢气受激拉曼位移管获得共振拉曼光谱实验所用的激发波长。为保证实验过程中样品的新鲜性,样品通过蠕动泵输送。溶液放置于150mL锥型瓶中,经导管在循环泵抽送下输送至喷嘴成液膜状流出,然后再经导管流回锥型瓶。拉曼信号的检测由CCD检测器完成。每个样品拉曼信号的检测时间为60~150s,作为一次输出数据。采集20~30次 (检测时间及采集次数由样品谱图的性噪比来确定),最后得到共振拉曼光谱。通过共振拉曼实验中获取的纯溶剂拉曼谱图,使用origin软件包完成对溶液共振拉曼光谱中溶剂峰的扣减,得到5个激发波长下PPD分别在环己烷和乙腈溶剂中的共振拉曼光谱。具体校正通过Origin软件的自编程序完成。
(2)理论计算:
所有量子化学计算均由Gaussian 09W程序包完成。采用密度泛函理论 (DFT) 和含时密度泛函理论 (TD-DFT) 方法,在B3LYP/6-311++G (d,p) 计算水平下获得PPD的几何结构和简正振动频率,在B3LYP-TD/6-311++G (d,p) 计算水平得到PPD的电子跃迁能。
(3)结论:
a) 气相条件下,PPD的稳定构型是非平面型的,属于C1点群。理论计算的优化结果和傅里叶拉曼实验的结果的对比证实了这一点。
b) PPD的电子吸收光谱表明,其电子吸收的跃迁主体是π→π*的跃迁,归属为A-电子吸收带,该吸收带对S4态Franck-Condon区域激发态结构有主要作用。
c) A-电子吸收带的共振拉曼光谱可以用于识别C=O的伸缩振动、环上CC的伸缩振动、环上CH的摇摆振动、甲基的面内摇摆振动以及C=O的面外变形振动。其中C=O伸缩振动、环上CC的伸缩振动和环上CH的面内摇摆的基频、倍频和组合频占据了共振拉曼光谱强度的绝大部分,这表明A-带Franck-Condon区域激发态结构动力学主要沿着C=O伸缩振动和环上CC的伸缩振动以及环上CH的面内摇摆等反应坐标展开。
[1]樊瑞雪,薛佳丹,万军民等.1-苯基-1,2-丙二酮的振动光谱和理论计算研究[J].浙江理工大学学报,2014,31(07):481-486.
了解1-苯基-1,2-丙二酮关于振动光谱和理论计算的相关研究,对于正确使用和认识其潜在风险非常重要。
背景:1-苯基-1,2-丙二酮(1-phenyl-1,2-propanedione,PPD)是一种典型的α-二羰基化合物,除了用于不对称有机合成反应外,还可用作光致聚合牙齿树脂复合材料的光敏剂。Leonard等人研究了PPD分子n-π*的电子吸收光谱,发现扭转角度对PPD的吸收有很大影响。SusyLopes等人研究了低温基质隔离下PPD的分子异构化以及紫外诱发的PPD去羰基化现象,进一步探讨了PPD的光化学和光物理现象。
振动光谱和理论计算研究:
利用共振拉曼光谱技术结合密度泛函理论计算,研究了PPD的激发态结构动力学,通过分析振动光谱和电子吸收光谱,考察PPD在激发态时分子结构以及电子振动耦合吸收等方面的信息。
(1)实验方法:
分别以环己烷和乙腈做溶剂,配制PPD溶液的浓度约为3.00~4.00 mmol/L。通过四倍频激光线 (波长266nm) 和氢气受激拉曼位移管获得共振拉曼光谱实验所用的激发波长。为保证实验过程中样品的新鲜性,样品通过蠕动泵输送。溶液放置于150mL锥型瓶中,经导管在循环泵抽送下输送至喷嘴成液膜状流出,然后再经导管流回锥型瓶。拉曼信号的检测由CCD检测器完成。每个样品拉曼信号的检测时间为60~150s,作为一次输出数据。采集20~30次 (检测时间及采集次数由样品谱图的性噪比来确定),最后得到共振拉曼光谱。通过共振拉曼实验中获取的纯溶剂拉曼谱图,使用origin软件包完成对溶液共振拉曼光谱中溶剂峰的扣减,得到5个激发波长下PPD分别在环己烷和乙腈溶剂中的共振拉曼光谱。具体校正通过Origin软件的自编程序完成。
(2)理论计算:
所有量子化学计算均由Gaussian 09W程序包完成。采用密度泛函理论 (DFT) 和含时密度泛函理论 (TD-DFT) 方法,在B3LYP/6-311++G (d,p) 计算水平下获得PPD的几何结构和简正振动频率,在B3LYP-TD/6-311++G (d,p) 计算水平得到PPD的电子跃迁能。
(3)结论:
a) 气相条件下,PPD的稳定构型是非平面型的,属于C1点群。理论计算的优化结果和傅里叶拉曼实验的结果的对比证实了这一点。
b) PPD的电子吸收光谱表明,其电子吸收的跃迁主体是π→π*的跃迁,归属为A-电子吸收带,该吸收带对S4态Franck-Condon区域激发态结构有主要作用。
c) A-电子吸收带的共振拉曼光谱可以用于识别C=O的伸缩振动、环上CC的伸缩振动、环上CH的摇摆振动、甲基的面内摇摆振动以及C=O的面外变形振动。其中C=O伸缩振动、环上CC的伸缩振动和环上CH的面内摇摆的基频、倍频和组合频占据了共振拉曼光谱强度的绝大部分,这表明A-带Franck-Condon区域激发态结构动力学主要沿着C=O伸缩振动和环上CC的伸缩振动以及环上CH的面内摇摆等反应坐标展开。
[1]樊瑞雪,薛佳丹,万军民等.1-苯基-1,2-丙二酮的振动光谱和理论计算研究[J].浙江理工大学学报,2014,31(07):481-486.
1
本文将讲述关于4-氟苯乙烯用于金属锂-碳纳米管的表面修饰时有何作用,旨在为相关领域的研究人员提供参考。
简述:4-氟苯乙烯,英文名称:4-Fluorostyrene,CAS:405-99-2,分子式:C8H7F,外观与性状:无色液体。4-氟苯乙烯是一种苯乙烯类化合物,具有丰富的化学转化性质。其不饱和双键结构可进行多种官能团转化反应,包括亲核加成反应、硼氢化反应等。因此,在基础有机化学研究中有着广泛的应用。
应用:对Li-CNT进行表面修饰。
金属锂负极由于比容量高(3860 mAh·g?1)及氧化还原电位极低(?3.04 V vs. 标准氢气电极(SHE)),被认为是实现高能量密度锂电池的理想负极。然而,金属锂电极与电解液反应剧烈,且锂离子在电极表面沉积不均匀容易产生枝晶,导致其循环稳定性和安全性都较差,限制了其应用推广。
刘亚等人前期通过构建金属锂-碳纳米管(Li-CNT)复合结构,极大的提高了金属锂的比表面积,降低了电极电流密度,从而有效地抑制了锂枝晶的生长,提高了金属锂电极的循环稳定性和安全性能。在前期工作基础上,采用简单的液相反应,利用4-氟苯乙烯(FPS)对Li-CNT进行表面修饰并进行原位聚合,得到了表面富含氟化锂(Li F)保护层的Li-CNT(FPS-Li-CNT)。材料制备与电极极片制备如下:
(1)Li-CNT是按照熔融浸渍法进行制备。首先通过喷雾干燥技术制备出碳纳米管球,再将4 g的金属锂块与2 g的碳纳米管微球混合在热反应加热器当中,200℃下机械搅拌20 min,转速500 r·min?1,搅拌完成后冷却至室温,得到的Li-CNT。表面修饰实验流程如图1a示意图所示,取FPS溶解于无水四氢呋喃(THF,纯度> 99.9%)中,得到浓度为0.1%质量比的混合溶液。取1 g的锂碳复合微球加入到10 g的混合溶液中,磁力搅拌10 min之后,溶液经过抽滤之后得到的样品在50℃烘箱中干燥12 h,得到目标产物(FPS-Li-CNT)。该制备过程在氩气保护氛围的手套箱中进行。
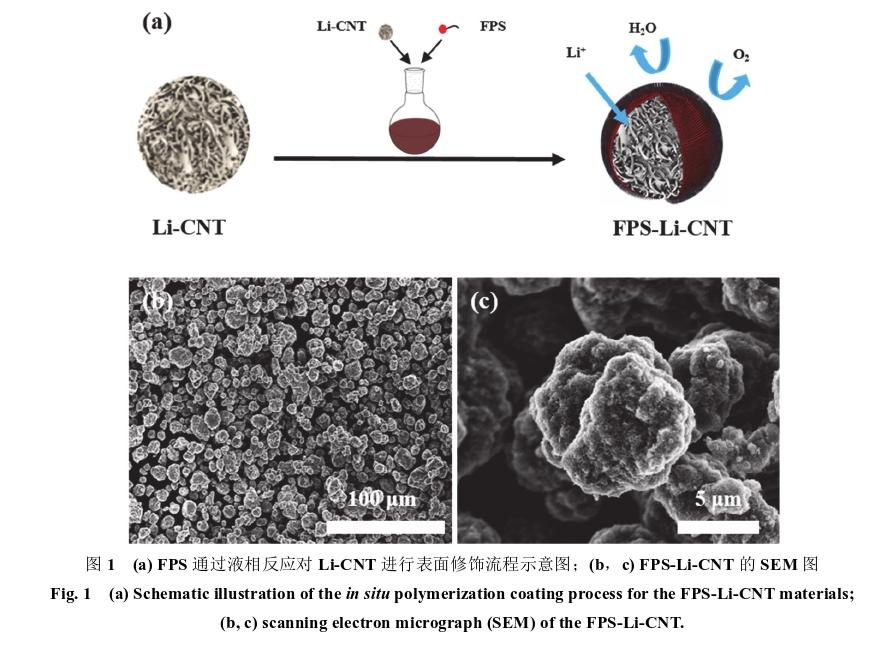
(2)FPS-Li-CNT||FPS-Li-CNT与Li-CNT||Li-CNT 对称电池测试所用极片,是将样品压在泡沫铜制作成直径为15 mm的极片,极片组成对称电池进行 电化学相关测试。组装成全电池的负极极片,我们 是采用混浆涂布工艺制备的。混浆涂布工艺如下:取320 mg的FPS-Li-CNT样品,40 mg的乙炔黑(AB) 作为导电剂,20 mg的聚苯乙烯(PS,相对分子质量 2000000)与20 mg的丁苯橡胶(SBR)作为粘结剂,溶于1.5 mL对二甲苯溶液 中,磁力搅拌12 h,浆料充分混合后,利用刮刀涂布于铜箔上,涂布刮刀厚度250 μm,真空烘干12 h,即得到FPS-Li-CNT负极极片。该制备过程在氩气保护氛围的手套箱中进行。
正极选择的是磷酸铁锂(LFP)。取45 mg PVDF作为粘结剂、45 mg乙炔黑(AB)作为导电剂、510 mg的LFP 粉体混合在1.5 mL的N-甲基吡咯烷酮(NMP)中,磁力搅拌12 h,混合均匀后,利用刮刀在铝箔上进行涂布,涂布厚度为275 μm,110℃条件下干燥12 h,得到LFP极片,面容量1.2 mAh·cm?2。
结论:该表面修饰层能够有效抑制电解液和空气对Li-CNT的侵蚀,显著的提高了Li CNT电极的界面稳定性。FPS-Li-CNT与磷酸铁锂正极(LFP)组成的LFP||FPS-Li-CNT全电池,在正负极容量配比为1 : 6条件下,能够稳定循环280圈,库伦效率达到97.7%。
参考文献:
[1]刘亚,郑磊,谷巍等.原位聚合表面修饰的金属锂负极[J].物理化学学报,2021,37(01):124-131.
显示全部本文将讲述关于4-氟苯乙烯用于金属锂-碳纳米管的表面修饰时有何作用,旨在为相关领域的研究人员提供参考。
简述:4-氟苯乙烯,英文名称:4-Fluorostyrene,CAS:405-99-2,分子式:C8H7F,外观与性状:无色液体。4-氟苯乙烯是一种苯乙烯类化合物,具有丰富的化学转化性质。其不饱和双键结构可进行多种官能团转化反应,包括亲核加成反应、硼氢化反应等。因此,在基础有机化学研究中有着广泛的应用。
应用:对Li-CNT进行表面修饰。
金属锂负极由于比容量高(3860 mAh·g?1)及氧化还原电位极低(?3.04 V vs. 标准氢气电极(SHE)),被认为是实现高能量密度锂电池的理想负极。然而,金属锂电极与电解液反应剧烈,且锂离子在电极表面沉积不均匀容易产生枝晶,导致其循环稳定性和安全性都较差,限制了其应用推广。
刘亚等人前期通过构建金属锂-碳纳米管(Li-CNT)复合结构,极大的提高了金属锂的比表面积,降低了电极电流密度,从而有效地抑制了锂枝晶的生长,提高了金属锂电极的循环稳定性和安全性能。在前期工作基础上,采用简单的液相反应,利用4-氟苯乙烯(FPS)对Li-CNT进行表面修饰并进行原位聚合,得到了表面富含氟化锂(Li F)保护层的Li-CNT(FPS-Li-CNT)。材料制备与电极极片制备如下:
(1)Li-CNT是按照熔融浸渍法进行制备。首先通过喷雾干燥技术制备出碳纳米管球,再将4 g的金属锂块与2 g的碳纳米管微球混合在热反应加热器当中,200℃下机械搅拌20 min,转速500 r·min?1,搅拌完成后冷却至室温,得到的Li-CNT。表面修饰实验流程如图1a示意图所示,取FPS溶解于无水四氢呋喃(THF,纯度> 99.9%)中,得到浓度为0.1%质量比的混合溶液。取1 g的锂碳复合微球加入到10 g的混合溶液中,磁力搅拌10 min之后,溶液经过抽滤之后得到的样品在50℃烘箱中干燥12 h,得到目标产物(FPS-Li-CNT)。该制备过程在氩气保护氛围的手套箱中进行。
(2)FPS-Li-CNT||FPS-Li-CNT与Li-CNT||Li-CNT 对称电池测试所用极片,是将样品压在泡沫铜制作成直径为15 mm的极片,极片组成对称电池进行 电化学相关测试。组装成全电池的负极极片,我们 是采用混浆涂布工艺制备的。混浆涂布工艺如下:取320 mg的FPS-Li-CNT样品,40 mg的乙炔黑(AB) 作为导电剂,20 mg的聚苯乙烯(PS,相对分子质量 2000000)与20 mg的丁苯橡胶(SBR)作为粘结剂,溶于1.5 mL对二甲苯溶液 中,磁力搅拌12 h,浆料充分混合后,利用刮刀涂布于铜箔上,涂布刮刀厚度250 μm,真空烘干12 h,即得到FPS-Li-CNT负极极片。该制备过程在氩气保护氛围的手套箱中进行。
正极选择的是磷酸铁锂(LFP)。取45 mg PVDF作为粘结剂、45 mg乙炔黑(AB)作为导电剂、510 mg的LFP 粉体混合在1.5 mL的N-甲基吡咯烷酮(NMP)中,磁力搅拌12 h,混合均匀后,利用刮刀在铝箔上进行涂布,涂布厚度为275 μm,110℃条件下干燥12 h,得到LFP极片,面容量1.2 mAh·cm?2。
结论:该表面修饰层能够有效抑制电解液和空气对Li-CNT的侵蚀,显著的提高了Li CNT电极的界面稳定性。FPS-Li-CNT与磷酸铁锂正极(LFP)组成的LFP||FPS-Li-CNT全电池,在正负极容量配比为1 : 6条件下,能够稳定循环280圈,库伦效率达到97.7%。
参考文献:
[1]刘亚,郑磊,谷巍等.原位聚合表面修饰的金属锂负极[J].物理化学学报,2021,37(01):124-131.
1
本文将介绍如何测定2,2-二噻吩基乙醇酸甲酯的含量,旨在为药物噻托溴铵的使用及相关分析化学提供实验支持。
背景:2,2-二噻吩基乙醇酸甲酯,英文名称:methyl 2-hydroxy-2,2-dithiophen-2-ylacetate,CAS:26447-85-8,分子式:C11H10O3S2。2,2-二噻吩基乙醇酸甲酯是生产长效支气管扩张剂噻托溴铵的中间体。
噻托溴铵(tiotropium bromide)是一种抗胆碱药物,由德国柏林格殷格翰(Boehringer Ingelheim)公司研发,与辉瑞(Pfizer)公司共同销售,2 002年6月首次在荷兰和菲律宾上市,主要用于慢性阻塞性肺疾病(COPD)的维持治疗。噻托溴铵与M1和M3受体解离缓慢,能在较长的时间内阻滞胆碱能神经介导的支气管平滑肌收缩,而与M2受体解离速度比较快,故其对支气管平滑肌松弛有良好的选择性,治疗慢性阻塞性肺疾病患者效果显著。
在制备噻托溴铵的过程中,主要有以下几种反应中间体:东莨菪醇、二(2-噻吩基)乙醇酸东莨菪酯和2,2-二噻吩基乙醇酸甲酯。
测定:
陈红等人建立了测定二(2-噻吩基)乙醇酸东莨菪酯和2,2-二噻吩基乙醇酸甲酯的含量的HPLC方法。采用μBondapakTMC18液相色谱柱(3.9mm i.d.×250mm,10μm),以乙腈/戊烷磺酸钠溶液(35/65,v/v,pH=7.0)为流动相,流速设定为0.8mL.min-1,检测波长为215nm时,实现了这两种酯的良好分离分析。实验方法如下:
1. 溶液的配制
(1)混合标准溶液的配制
准确称取0.004 85 g二(2-噻吩基)乙醇酸东莨菪酯和0.005 15 g 2,2-二噻吩基乙醇酸甲酯,置于25 mL容量瓶中,然后加入70%甲醇水溶液,超声溶解后定容,摇匀,二(2-噻吩基)乙醇酸东莨菪酯储备液浓度为194 μg·mL-1,2,2-二噻吩基乙醇酸甲酯储备液浓度为206 μg·mL-1。
(2)离子对试剂溶液的配制
准确称取0.696 0 g戊烷磺酸钠,置于烧杯中,加入400 mL二次蒸馏水,完全溶解后,用磷酸调节pH至7.0,经G4砂芯漏斗过滤后,超声脱气10 min,配制得到10 mmol·L-1 浓度的离子对试剂溶液。
2. 色谱条件
色谱柱:μBondapakTMC18 液相色谱柱(3.9 mm i.d.×250 mm,10 μm);流动相:乙腈/戊烷磺酸钠溶液(35/65,v/v);柱温:25 ℃;流速:0.8 mL·min-1;进样量:10 μL;检测波长:215 nm;流动相经G4砂芯漏斗过滤后,超声脱气10 min。
结果为:二(2-噻吩基)乙醇酸东莨菪酯的线性范围为1.52~194μg.mL-1,r=0.999 6;2,2-二噻吩基乙醇酸甲酯的线性范围为1.61~206μg.mL-1,r=0.999 6。平均加标回收率及RSD分别为94.43%(RSD 5.66%)和96.21%(RSD 4.39%)。本方法灵敏度高、选择性好、操作简单,可方便地用于监测噻托溴铵合成反应的过程,也适用于检测噻托溴铵中间体含量。
参考文献:
[1]陈红,李来生,方奕珊等.HPLC法测定二(2-噻吩基)乙醇酸东莨菪酯和2,2-二噻吩基乙醇酸甲酯的含量[J].南昌大学学报(理科版),2011,35(05):447-449+463.
显示全部本文将介绍如何测定2,2-二噻吩基乙醇酸甲酯的含量,旨在为药物噻托溴铵的使用及相关分析化学提供实验支持。
背景:2,2-二噻吩基乙醇酸甲酯,英文名称:methyl 2-hydroxy-2,2-dithiophen-2-ylacetate,CAS:26447-85-8,分子式:C11H10O3S2。2,2-二噻吩基乙醇酸甲酯是生产长效支气管扩张剂噻托溴铵的中间体。
噻托溴铵(tiotropium bromide)是一种抗胆碱药物,由德国柏林格殷格翰(Boehringer Ingelheim)公司研发,与辉瑞(Pfizer)公司共同销售,2 002年6月首次在荷兰和菲律宾上市,主要用于慢性阻塞性肺疾病(COPD)的维持治疗。噻托溴铵与M1和M3受体解离缓慢,能在较长的时间内阻滞胆碱能神经介导的支气管平滑肌收缩,而与M2受体解离速度比较快,故其对支气管平滑肌松弛有良好的选择性,治疗慢性阻塞性肺疾病患者效果显著。
在制备噻托溴铵的过程中,主要有以下几种反应中间体:东莨菪醇、二(2-噻吩基)乙醇酸东莨菪酯和2,2-二噻吩基乙醇酸甲酯。
测定:
陈红等人建立了测定二(2-噻吩基)乙醇酸东莨菪酯和2,2-二噻吩基乙醇酸甲酯的含量的HPLC方法。采用μBondapakTMC18液相色谱柱(3.9mm i.d.×250mm,10μm),以乙腈/戊烷磺酸钠溶液(35/65,v/v,pH=7.0)为流动相,流速设定为0.8mL.min-1,检测波长为215nm时,实现了这两种酯的良好分离分析。实验方法如下:
1. 溶液的配制
(1)混合标准溶液的配制
准确称取0.004 85 g二(2-噻吩基)乙醇酸东莨菪酯和0.005 15 g 2,2-二噻吩基乙醇酸甲酯,置于25 mL容量瓶中,然后加入70%甲醇水溶液,超声溶解后定容,摇匀,二(2-噻吩基)乙醇酸东莨菪酯储备液浓度为194 μg·mL-1,2,2-二噻吩基乙醇酸甲酯储备液浓度为206 μg·mL-1。
(2)离子对试剂溶液的配制
准确称取0.696 0 g戊烷磺酸钠,置于烧杯中,加入400 mL二次蒸馏水,完全溶解后,用磷酸调节pH至7.0,经G4砂芯漏斗过滤后,超声脱气10 min,配制得到10 mmol·L-1 浓度的离子对试剂溶液。
2. 色谱条件
色谱柱:μBondapakTMC18 液相色谱柱(3.9 mm i.d.×250 mm,10 μm);流动相:乙腈/戊烷磺酸钠溶液(35/65,v/v);柱温:25 ℃;流速:0.8 mL·min-1;进样量:10 μL;检测波长:215 nm;流动相经G4砂芯漏斗过滤后,超声脱气10 min。
结果为:二(2-噻吩基)乙醇酸东莨菪酯的线性范围为1.52~194μg.mL-1,r=0.999 6;2,2-二噻吩基乙醇酸甲酯的线性范围为1.61~206μg.mL-1,r=0.999 6。平均加标回收率及RSD分别为94.43%(RSD 5.66%)和96.21%(RSD 4.39%)。本方法灵敏度高、选择性好、操作简单,可方便地用于监测噻托溴铵合成反应的过程,也适用于检测噻托溴铵中间体含量。
参考文献:
[1]陈红,李来生,方奕珊等.HPLC法测定二(2-噻吩基)乙醇酸东莨菪酯和2,2-二噻吩基乙醇酸甲酯的含量[J].南昌大学学报(理科版),2011,35(05):447-449+463.
1
本文将讲述2,2'-联喹啉-4,4'-二羧酸二钠如何应用于Cu的分析测定,以期为分析化学,环境安全等领域的的研究人员提供实验支持。
简述:2,2'-联喹啉-4,4'-二羧酸二钠,英文名为Bicinchoninic Acid Disodium Salt。2,2'-联喹啉-4,4'-二羧酸二钠属于羧酸类衍生物,可用作医药中间体,也可用于Cu和蛋白的分析测定。
应用:
1. 测定循环水中微量铜
工业循环冷却水中铜离子的测定一般采用原子吸收法(GB/T 14638.2-1993)和二乙基二硫代氨基甲 酸钠比色法(GB/T 14418-1993,前者需要昂贵的 原子吸收光谱仪,后者需要有机溶剂萃取等繁杂操作。
为了提高检测效率,严琳等人利用BCA(2,2’-联喹啉-4,4’-二甲酸二钠盐) 在碱性条件下能特异性的与亚铜离子形成紫色Cu(I)-BCA配合物,并与铜离子浓度有良好的线性关系的原理,建立一种快捷、准确的比色测定法。测试方法为:
(1)BCA比色法:取两支50 mL比色管,一支准确移取适量的铜标准溶液,一支不加,作为空白对照,两支比色管中都加入试剂B、试剂A,再用去离子水稀释至刻度,充分摇匀后静置,待显色完全;用5 cm比色皿于一定波长处测量溶液的吸光度A,确定最适波长、pH、试剂用量以及显色时间,并绘制最佳实验条件下的铜含量回归曲线。用循环水样品代替铜标准溶液,采用上述方法测定铜含量。
(2)二乙基二硫代氨基甲酸钠比色法:按照 GB/T 14418-1993进行测定。
结论:在盐酸羟胺存在的条件下,pH=10.0缓冲介质中,铜(I)与BCA形成紫色配合物λmax=562 nm,E562=7.68×103 L·mol-1·cm-1,铜含量在0~1 μg·mL-1内符合比尔定律。该方法稳定准确,能够满足生产检测需求。
2. 测定食品中的微量铜
饮用水及食品中均含有一定量的铜,但如果人体摄入的铜过量将对人体构成危害。近年由于农业上大量使用含铜杀虫剂和除霉剂,加上工业“三废”的污染,使许多食品中铜含量超标。因此,食品中铜的允许限量一般不超过5~20mg/kg。测定食品中铜的含量是食品检验中的一项重要工作。
刘璇等人研究BCA(2,2'-联喹啉-4,4'-二甲酸二钠盐)与铜的显色反应,建立测量食品微量铜的光度分析方法。在盐酸羟胺存在条件下,pH11.0缓冲介质中,BCA试剂与铜在物质的量的比为2:1的条件下反应生成的紫色络合物, λmax=562nm,ε562nm=7.72×103L/(mol·cm),铜含量在0~4.8μg/mL内符合比尔定律;经测定茶叶和海带中的铜含量,结果与原子吸收法的测定结果相符。结果表明:该方法稳定准确,可用于食品中微量铜的测定。测定方法具体为:
(1)BCA比色法:于25mL容量瓶中加入一定量的铜标准液,然后加入3.00mL盐酸羟胺溶液,振荡;再加入 5.00mL BCA 试剂,用水稀释至刻度,充分摇匀后放置 15min,待显色完全后在分光光度计上,以相应的试剂空白(用等量的二次蒸馏水代替铜标准液,其他试剂加入同上)作参比,用1cm比色皿于562nm处测量溶液的吸光度,以此进行铜的定量分析。
(2)原子吸收法:按照GB/T 5009.13—2003《食品中铜的测定方法》进行。测定条件:火焰类型:乙炔/ 空气;测定波长324.7 5nm(铜空心阴极灯);灯电流 2 mA;狭缝0.2 n m。
参考文献:
[1]严琳,陈达,司有银. BCA比色法测定循环水中微量铜的优化 [J]. 现代食品, 2018, (23): 138-141. DOI:10.16736/j.cnki.cn41-1434/ts.2018.23.040.
[2]刘璇,冯志明. BCA法测定食品中的微量铜 [J]. 食品科学, 2011, 32 (14): 277-280.
显示全部本文将讲述2,2'-联喹啉-4,4'-二羧酸二钠如何应用于Cu的分析测定,以期为分析化学,环境安全等领域的的研究人员提供实验支持。
简述:2,2'-联喹啉-4,4'-二羧酸二钠,英文名为Bicinchoninic Acid Disodium Salt。2,2'-联喹啉-4,4'-二羧酸二钠属于羧酸类衍生物,可用作医药中间体,也可用于Cu和蛋白的分析测定。
应用:
1. 测定循环水中微量铜
工业循环冷却水中铜离子的测定一般采用原子吸收法(GB/T 14638.2-1993)和二乙基二硫代氨基甲 酸钠比色法(GB/T 14418-1993,前者需要昂贵的 原子吸收光谱仪,后者需要有机溶剂萃取等繁杂操作。
为了提高检测效率,严琳等人利用BCA(2,2’-联喹啉-4,4’-二甲酸二钠盐) 在碱性条件下能特异性的与亚铜离子形成紫色Cu(I)-BCA配合物,并与铜离子浓度有良好的线性关系的原理,建立一种快捷、准确的比色测定法。测试方法为:
(1)BCA比色法:取两支50 mL比色管,一支准确移取适量的铜标准溶液,一支不加,作为空白对照,两支比色管中都加入试剂B、试剂A,再用去离子水稀释至刻度,充分摇匀后静置,待显色完全;用5 cm比色皿于一定波长处测量溶液的吸光度A,确定最适波长、pH、试剂用量以及显色时间,并绘制最佳实验条件下的铜含量回归曲线。用循环水样品代替铜标准溶液,采用上述方法测定铜含量。
(2)二乙基二硫代氨基甲酸钠比色法:按照 GB/T 14418-1993进行测定。
结论:在盐酸羟胺存在的条件下,pH=10.0缓冲介质中,铜(I)与BCA形成紫色配合物λmax=562 nm,E562=7.68×103 L·mol-1·cm-1,铜含量在0~1 μg·mL-1内符合比尔定律。该方法稳定准确,能够满足生产检测需求。
2. 测定食品中的微量铜
饮用水及食品中均含有一定量的铜,但如果人体摄入的铜过量将对人体构成危害。近年由于农业上大量使用含铜杀虫剂和除霉剂,加上工业“三废”的污染,使许多食品中铜含量超标。因此,食品中铜的允许限量一般不超过5~20mg/kg。测定食品中铜的含量是食品检验中的一项重要工作。
刘璇等人研究BCA(2,2'-联喹啉-4,4'-二甲酸二钠盐)与铜的显色反应,建立测量食品微量铜的光度分析方法。在盐酸羟胺存在条件下,pH11.0缓冲介质中,BCA试剂与铜在物质的量的比为2:1的条件下反应生成的紫色络合物, λmax=562nm,ε562nm=7.72×103L/(mol·cm),铜含量在0~4.8μg/mL内符合比尔定律;经测定茶叶和海带中的铜含量,结果与原子吸收法的测定结果相符。结果表明:该方法稳定准确,可用于食品中微量铜的测定。测定方法具体为:
(1)BCA比色法:于25mL容量瓶中加入一定量的铜标准液,然后加入3.00mL盐酸羟胺溶液,振荡;再加入 5.00mL BCA 试剂,用水稀释至刻度,充分摇匀后放置 15min,待显色完全后在分光光度计上,以相应的试剂空白(用等量的二次蒸馏水代替铜标准液,其他试剂加入同上)作参比,用1cm比色皿于562nm处测量溶液的吸光度,以此进行铜的定量分析。
(2)原子吸收法:按照GB/T 5009.13—2003《食品中铜的测定方法》进行。测定条件:火焰类型:乙炔/ 空气;测定波长324.7 5nm(铜空心阴极灯);灯电流 2 mA;狭缝0.2 n m。
参考文献:
[1]严琳,陈达,司有银. BCA比色法测定循环水中微量铜的优化 [J]. 现代食品, 2018, (23): 138-141. DOI:10.16736/j.cnki.cn41-1434/ts.2018.23.040.
[2]刘璇,冯志明. BCA法测定食品中的微量铜 [J]. 食品科学, 2011, 32 (14): 277-280.
1
本文将讲述4-二氟甲氧基-3-羟基苯甲醛中的杂质控制的方法,希望能够有效地控制罗氟司特的质量。
背景:罗氟司特(Roflumilast)中的主要杂质是主成分峰后的杂质峰(杂质A)。根据罗氟司特的合成路径,推测其产生路径与罗氟司特的起始原料二氟甲氧基-3-羟基苯甲醛(DFMHB)中微量含有的3,4-二羟基苯甲醛有关。该杂质与罗氟司特的性质非常相似,一旦产生则难以用常规手段去除。目前已建立了测定DFMHB中3,4-二羟基苯甲醛的HPLC方法,可有效地控制罗氟司特的质量。
张佳楠等人建立罗氟司特起始原料(4-二氟甲氧基-3-羟基苯甲醛)中的杂质控制方法。具体如下:
(1)色谱条件
采用VisionHT C18 色谱柱(250 mm×4.6 mm,5μm),流动相为甲醇-0.01 mol· L-1 磷酸二氢钾溶液(40∶60),流速1.0 mL·min-1 ,检测波长254 nm,柱温30℃,进样体积20μL。
(2)溶液的制备
精取约12.5 mg DFMHB,置25 mL量瓶中,加流动相溶解并定容,制成约 0.5 mg·mL-1 供试品溶液。精密量取1 mL该溶液,置100 mL量瓶中,并以流动相定容,再精密量取 1 mL稀释溶液,置10 mL量瓶中,以流动相定容,制 成约0.5μg·mL-1 对照品溶液。精密称取约 12.5 mg DHBA,置25 mL量瓶中,加流动相溶解并定容,作为杂质贮备液,再精密量取1 mL杂质贮备液,置100 mL量瓶中,并以流动相定容,制成约 5μg·mL-1 杂质对照品溶液。
(3)系统适应性试验
精密量取1 mL供试品溶 液与0.1 mL杂质贮备液,置同一100 mL量瓶中,用流动相定容,作为系统适用性试液。按(1)项 色谱条件进样,记录色谱图。上述两种物质色谱峰的峰形良好,均能达到基线分离。
(4)线性关系及定量限的考察
精密量取0.2、 0.4、0.6、0.8、1.0、1.2 mL杂质贮备液,分别置100 mL量瓶中,用流动相定容,即得系列标准溶液,按(1)项色谱条件,进样分析,以样品浓度(μg· mL-1 )对峰面积进行线性回归,得线性回归方程为:Y=4.37×104X-3.59×102 ,1.028~6.168μg· mL-1 DHBA与峰面积的线性关系良好(r=0.9994,n=6)。取杂质对照品溶液,用逐步稀释法测定检测限(S/N≥3)与定量限(S/N≥10),结果显示: DHBA的检测限为0.1028 ng,定量限为0.3074 ng,该定量限与检测限可满足DHBA的检测要求。
(5)精密度与稳定性试验
取杂质对照品溶液,按(1)项色谱条件,连续进样6次,计算得DH BA峰面积的RSD=0.31%,表明仪器的精密度良好。精密量取0.5 mL杂质贮备液,置50 mL量瓶中,以流动相定容。分别于0、2、4、6、8 h时进样分析。8 h后DHBA的增加量为0.11%。
(6加样回收试验
精密称取约50 mg DFMHB 共9份,分别置100 mL量瓶中,分别精密加入0.8、1.0、1.2 mL杂质贮备液,每个浓度制备3份,并用流动相定容,作为低、中、高浓度的供试品溶液。取供试品溶液各5 mL,分别置10 mL量瓶中并以流动相定容,再将稀释的溶液按“1.2.2”项色谱条件进样分析。以峰面积按外标法计算其含量。可得DH BA加样回收率均值为99.82%,RSD=0.54%。
(7)样品的测定
取3批样品,按(1)色谱条件,测定DFMHB中DHBA的含量。计算3批样品中的杂质含量分别为:未检出、1.8‰、0.5‰
结果:样品的线性范围为0.1028~0.6168μg·ml-1 (r=0.999),平均回收率为99.75%,RSD=0.57%。所用方法能有效地控制4-二氟甲氧基-3-羟基苯甲醛中的3,4-二羟基苯甲醛杂质,可得到高纯度的罗氟司特。
参考文献:
[1]张佳楠,严相平.HPLC测定罗氟司特起始原料中的3,4-二羟基苯甲醛[J].华西药学杂志,2016,31(06):634-635.DOI:10.13375/j.cnki.wcjps.2016.06.025.
显示全部本文将讲述4-二氟甲氧基-3-羟基苯甲醛中的杂质控制的方法,希望能够有效地控制罗氟司特的质量。
背景:罗氟司特(Roflumilast)中的主要杂质是主成分峰后的杂质峰(杂质A)。根据罗氟司特的合成路径,推测其产生路径与罗氟司特的起始原料二氟甲氧基-3-羟基苯甲醛(DFMHB)中微量含有的3,4-二羟基苯甲醛有关。该杂质与罗氟司特的性质非常相似,一旦产生则难以用常规手段去除。目前已建立了测定DFMHB中3,4-二羟基苯甲醛的HPLC方法,可有效地控制罗氟司特的质量。
张佳楠等人建立罗氟司特起始原料(4-二氟甲氧基-3-羟基苯甲醛)中的杂质控制方法。具体如下:
(1)色谱条件
采用VisionHT C18 色谱柱(250 mm×4.6 mm,5μm),流动相为甲醇-0.01 mol· L-1 磷酸二氢钾溶液(40∶60),流速1.0 mL·min-1 ,检测波长254 nm,柱温30℃,进样体积20μL。
(2)溶液的制备
精取约12.5 mg DFMHB,置25 mL量瓶中,加流动相溶解并定容,制成约 0.5 mg·mL-1 供试品溶液。精密量取1 mL该溶液,置100 mL量瓶中,并以流动相定容,再精密量取 1 mL稀释溶液,置10 mL量瓶中,以流动相定容,制 成约0.5μg·mL-1 对照品溶液。精密称取约 12.5 mg DHBA,置25 mL量瓶中,加流动相溶解并定容,作为杂质贮备液,再精密量取1 mL杂质贮备液,置100 mL量瓶中,并以流动相定容,制成约 5μg·mL-1 杂质对照品溶液。
(3)系统适应性试验
精密量取1 mL供试品溶 液与0.1 mL杂质贮备液,置同一100 mL量瓶中,用流动相定容,作为系统适用性试液。按(1)项 色谱条件进样,记录色谱图。上述两种物质色谱峰的峰形良好,均能达到基线分离。
(4)线性关系及定量限的考察
精密量取0.2、 0.4、0.6、0.8、1.0、1.2 mL杂质贮备液,分别置100 mL量瓶中,用流动相定容,即得系列标准溶液,按(1)项色谱条件,进样分析,以样品浓度(μg· mL-1 )对峰面积进行线性回归,得线性回归方程为:Y=4.37×104X-3.59×102 ,1.028~6.168μg· mL-1 DHBA与峰面积的线性关系良好(r=0.9994,n=6)。取杂质对照品溶液,用逐步稀释法测定检测限(S/N≥3)与定量限(S/N≥10),结果显示: DHBA的检测限为0.1028 ng,定量限为0.3074 ng,该定量限与检测限可满足DHBA的检测要求。
(5)精密度与稳定性试验
取杂质对照品溶液,按(1)项色谱条件,连续进样6次,计算得DH BA峰面积的RSD=0.31%,表明仪器的精密度良好。精密量取0.5 mL杂质贮备液,置50 mL量瓶中,以流动相定容。分别于0、2、4、6、8 h时进样分析。8 h后DHBA的增加量为0.11%。
(6加样回收试验
精密称取约50 mg DFMHB 共9份,分别置100 mL量瓶中,分别精密加入0.8、1.0、1.2 mL杂质贮备液,每个浓度制备3份,并用流动相定容,作为低、中、高浓度的供试品溶液。取供试品溶液各5 mL,分别置10 mL量瓶中并以流动相定容,再将稀释的溶液按“1.2.2”项色谱条件进样分析。以峰面积按外标法计算其含量。可得DH BA加样回收率均值为99.82%,RSD=0.54%。
(7)样品的测定
取3批样品,按(1)色谱条件,测定DFMHB中DHBA的含量。计算3批样品中的杂质含量分别为:未检出、1.8‰、0.5‰
结果:样品的线性范围为0.1028~0.6168μg·ml-1 (r=0.999),平均回收率为99.75%,RSD=0.57%。所用方法能有效地控制4-二氟甲氧基-3-羟基苯甲醛中的3,4-二羟基苯甲醛杂质,可得到高纯度的罗氟司特。
参考文献:
[1]张佳楠,严相平.HPLC测定罗氟司特起始原料中的3,4-二羟基苯甲醛[J].华西药学杂志,2016,31(06):634-635.DOI:10.13375/j.cnki.wcjps.2016.06.025.
1
建立可靠的3,5-二氯苯甲酰氯测定分析方法对于3,5-二氯苯甲酰氯的质量控制具有重要意义。
简述:3,5-二氯苯甲酰氯为无色液体或结晶,密度:1.498g/cm3,熔点:28℃,沸点:249.1℃。溶解性:易溶于二氯亚砜,乙醚,氯仿等有机溶剂,极易与水反应生成3,5—二氯苯甲酸,是一种重要的医药、农药和染料中间体。
测定:
1. 柱前衍生反相高效液相色谱法
郭秀君等人以无水甲醇作柱前衍生试剂,建立了反相高效液相色谱法间接测定3,5-二氯苯甲酰氯的新方法。实验方法如下:
(1)色谱条件
色谱柱:C18(150mmx4.6mm,5μm);流动相甲醇-水(85:15);流速:1.0mL/min;检测波长:288nm;进样量:20μL;柱温:室温。
(2)3,5-二氯苯甲酰氯标准溶液
精密称定0.30g标准品,用少量无水甲醇溶解后,定量转入100mL的容量瓶中,并用无水甲醇定容。该溶液的质量浓度为3.0mg/mL。
(3)实验方法
分别移取不同体积的3,5-二氯苯甲酰氯标准溶液于6只25mL容量瓶中,用流动相释至刻度在确定的色谱条件下进样20μL,按归一化法定量分别测定其峰面积并对数据作回归分析。移取适量的样品酯化溶液于25mL 容量瓶中并用流动相稀释至刻度,在同样条件下测定其峰面积根据回归方程计算3,5-二氯苯甲酰氯的质量浓度,按计量关系换算样品中3,5-二氯苯甲酰氯的含量。
实验结果表明,在流动相:甲醇-水(85:15),流速:1.0mL/min,检测波长:288nm的条件下,定量校正曲线具有良好的线性关系,相关系数(r)为0.9999。方法用于样品中3,5-二氯苯甲酰氯的测定,RSD=0.19%(n=6),加标回收率为99.9%~105%。
2. 荷移增敏分光光度法
贾文平等人建立用荷移增敏分光光度法测定3,5-二氯苯甲酰氯的方法。利用3,5-二氯苯甲酰氯与羟胺反应的产物(3,5-二氯-N-羟基苯甲酰胺,DCHA)与对苯醌在氯代十六烷基吡啶(CPC)介质中形成稳定络合物,在485nm波长处测定其吸光度。实验方法具体如下:
准确移取0.50mL3,5-二氯苯甲酰氯使用液于10mL比色管中,加入5.0 mL羟胺溶液,反应25min后,加入1.0mL对苯醌,6min后,再加人1.0mL氯代十六烷基吡啶,稍振荡后静置,并用无水乙醇定容。以试剂空白作参比,在波长485 nm处测定吸光度。同样条件下,以试样空白作参比测定样品溶液的吸光度。
结果表明,3,5-二氯苯甲酸乙酯的检出限为0.15mg/L,线性范围(0.30~4.00)mg/L,相关系数(r)为0.9992,样品测定的RSD为3.65%(n=6),回收率为97.3%~100.1%。该法简便快速,条件易控制,测定结果满意。
参考文献:
[1]牛彬波. 3,5-二氯苯甲酰氯的合成研究[D]. 河北工业大学, 2015.
[2]郭秀君,李芳,蒋华江等. 柱前衍生反相高效液相色谱法快速测定3,5-二氯苯甲酰氯 [J]. 浙江化工, 2007, (05): 24-25.
[3]贾文平,李芳,蒋华江等. 荷移增敏分光光度法间接测定3,5-二氯苯甲酰氯 [J]. 科学技术与工程, 2007, (04): 596-598.
显示全部建立可靠的3,5-二氯苯甲酰氯测定分析方法对于3,5-二氯苯甲酰氯的质量控制具有重要意义。
简述:3,5-二氯苯甲酰氯为无色液体或结晶,密度:1.498g/cm3,熔点:28℃,沸点:249.1℃。溶解性:易溶于二氯亚砜,乙醚,氯仿等有机溶剂,极易与水反应生成3,5—二氯苯甲酸,是一种重要的医药、农药和染料中间体。
测定:
1. 柱前衍生反相高效液相色谱法
郭秀君等人以无水甲醇作柱前衍生试剂,建立了反相高效液相色谱法间接测定3,5-二氯苯甲酰氯的新方法。实验方法如下:
(1)色谱条件
色谱柱:C18(150mmx4.6mm,5μm);流动相甲醇-水(85:15);流速:1.0mL/min;检测波长:288nm;进样量:20μL;柱温:室温。
(2)3,5-二氯苯甲酰氯标准溶液
精密称定0.30g标准品,用少量无水甲醇溶解后,定量转入100mL的容量瓶中,并用无水甲醇定容。该溶液的质量浓度为3.0mg/mL。
(3)实验方法
分别移取不同体积的3,5-二氯苯甲酰氯标准溶液于6只25mL容量瓶中,用流动相释至刻度在确定的色谱条件下进样20μL,按归一化法定量分别测定其峰面积并对数据作回归分析。移取适量的样品酯化溶液于25mL 容量瓶中并用流动相稀释至刻度,在同样条件下测定其峰面积根据回归方程计算3,5-二氯苯甲酰氯的质量浓度,按计量关系换算样品中3,5-二氯苯甲酰氯的含量。
实验结果表明,在流动相:甲醇-水(85:15),流速:1.0mL/min,检测波长:288nm的条件下,定量校正曲线具有良好的线性关系,相关系数(r)为0.9999。方法用于样品中3,5-二氯苯甲酰氯的测定,RSD=0.19%(n=6),加标回收率为99.9%~105%。
2. 荷移增敏分光光度法
贾文平等人建立用荷移增敏分光光度法测定3,5-二氯苯甲酰氯的方法。利用3,5-二氯苯甲酰氯与羟胺反应的产物(3,5-二氯-N-羟基苯甲酰胺,DCHA)与对苯醌在氯代十六烷基吡啶(CPC)介质中形成稳定络合物,在485nm波长处测定其吸光度。实验方法具体如下:
准确移取0.50mL3,5-二氯苯甲酰氯使用液于10mL比色管中,加入5.0 mL羟胺溶液,反应25min后,加入1.0mL对苯醌,6min后,再加人1.0mL氯代十六烷基吡啶,稍振荡后静置,并用无水乙醇定容。以试剂空白作参比,在波长485 nm处测定吸光度。同样条件下,以试样空白作参比测定样品溶液的吸光度。
结果表明,3,5-二氯苯甲酸乙酯的检出限为0.15mg/L,线性范围(0.30~4.00)mg/L,相关系数(r)为0.9992,样品测定的RSD为3.65%(n=6),回收率为97.3%~100.1%。该法简便快速,条件易控制,测定结果满意。
参考文献:
[1]牛彬波. 3,5-二氯苯甲酰氯的合成研究[D]. 河北工业大学, 2015.
[2]郭秀君,李芳,蒋华江等. 柱前衍生反相高效液相色谱法快速测定3,5-二氯苯甲酰氯 [J]. 浙江化工, 2007, (05): 24-25.
[3]贾文平,李芳,蒋华江等. 荷移增敏分光光度法间接测定3,5-二氯苯甲酰氯 [J]. 科学技术与工程, 2007, (04): 596-598.
1
本文将讲述如何测定卡维地洛片中的杂质4-环氧丙烷氧基咔唑,旨在为卡维地洛有关物质的检测和质量控制提供参考思路。
背景:4-环氧丙烷氧基咔唑,英文名称:4-Glycidyloxycarbazole,CAS:51997-51-4,分子式:C15H13NO2,外观与性状:淡黄色至近乎于白色粉末,密度:1.327g/cm3。4-环氧丙烷氧基咔唑是卡维地洛的合成中间体。
卡维地洛是一种α、β受体阻断剂,阻断受体的同时具有舒张血管的作用,用于治疗轻度及中度高血压和心力衰竭。卡维地洛的已知杂质有N-(2-羟基-3-(2-(甲氧基苯氧)-乙基)胺卡维地洛(杂质A)、卡维地洛双咔唑(杂质B)、N-甲基苯卡维地洛(杂质C)、4-环氧丙烷氧基咔唑(杂质D)、2-(2-甲氧基苯氧基)乙胺(杂质E)等。其中杂质D是中间体,杂质E是合成原料,并且能由卡维地洛的降解产生,其他杂质为工艺杂质,因此方法应控制降解杂质—杂质D、E。
测定卡维地洛片中的4-环氧丙烷氧基咔唑:
康铁纯等人建立反相高效液相色谱法(RP-HPLC)测定卡维地洛片有关物质。方法为:采用YMC-Pack Pro C8色谱柱(150 mm× 4.6 mm,5μm),以乙腈-0.02 mol·L-1 磷酸二氢钾溶液(用磷酸调pH 2.0)为流动相进行梯度洗脱,流速1.0 mL·min-1 ,柱温 30℃,检测波长为220 nm(杂质E)和240 nm(其他杂质)。具体实验方法如下:
(1)色谱条件
采用YMC-Pack Pro C8色谱柱(150 mm×4.6 mm, 5μm),以2.72 g·L- 1 磷酸二氢钾溶液(用磷酸调节 pH 2.0)为流动相A,乙腈为流动相B,梯度洗脱(0~ 5 min,20%B;5~20 min,20%B→60%B;20~30 min,60%B;30~30.1 min,60%B→20%B;30.1~35 min,20%B),流速1.0 mL·min-1 ,柱温30℃,检测波长 220 nm(杂质E)和240 nm(其他杂质),进样体积 10μL。
(2)溶液的制备
供试品溶液取本品细粉适量(约相当于卡维地洛25 mg),置50 mL量瓶中,加稀释液[流动相A-流动相B(70∶30)]使卡维地洛溶解并稀释至刻度,摇匀,滤过,取续滤液即得。
对照品溶液取杂质D、E和卡维地洛的对照品各适量,精密称定,加稀释液溶解并稀释制成每1 mL 中各约含0.5μg的混合溶液,即得,色谱图见图2。
结果:卡维地洛与已知杂质及强制破坏实验产生的降解产物均能获得良好的分离,杂质D(4-环氧丙烷氧基咔唑)与杂质E[2-(2-甲氧基苯氧基)乙胺]质量浓度分别在0.063 98~ 12.80μg·mL-1 (r=0.999 9)和0.089 33~8.933μg·mL-1 (r=1.000)范围内与峰面积呈良好的线性关系。杂质D高、中、低浓度的回收率(n=3)分别为98.6%、96.5%、94.2%,RSD分别为1.1%、0.8%、1.4%;杂质E高、中、低浓度的回收率(n=3) 分别为99.2%、86%、92.7%,RSD分别为0.15%、0.65%、9.5%。杂质D和杂质E精密度试验的RSD(n=6)分别为1.4%和 0.31%,建立的方法与现行中国药典收载的方法相比能分离检测到更多的杂质。该方法经方法学验证,可用于卡维地洛有关物质的检测和质量控制。
参考文献:
[1]康铁纯,顾宵,何佳佳等.RP-HPLC测定卡维地洛片中的杂质D和杂质E[J].药物分析杂志,2015,35(10):1838-1842.DOI:10.16155/j.0254-1793.2015.10.26.
显示全部本文将讲述如何测定卡维地洛片中的杂质4-环氧丙烷氧基咔唑,旨在为卡维地洛有关物质的检测和质量控制提供参考思路。
背景:4-环氧丙烷氧基咔唑,英文名称:4-Glycidyloxycarbazole,CAS:51997-51-4,分子式:C15H13NO2,外观与性状:淡黄色至近乎于白色粉末,密度:1.327g/cm3。4-环氧丙烷氧基咔唑是卡维地洛的合成中间体。
卡维地洛是一种α、β受体阻断剂,阻断受体的同时具有舒张血管的作用,用于治疗轻度及中度高血压和心力衰竭。卡维地洛的已知杂质有N-(2-羟基-3-(2-(甲氧基苯氧)-乙基)胺卡维地洛(杂质A)、卡维地洛双咔唑(杂质B)、N-甲基苯卡维地洛(杂质C)、4-环氧丙烷氧基咔唑(杂质D)、2-(2-甲氧基苯氧基)乙胺(杂质E)等。其中杂质D是中间体,杂质E是合成原料,并且能由卡维地洛的降解产生,其他杂质为工艺杂质,因此方法应控制降解杂质—杂质D、E。
测定卡维地洛片中的4-环氧丙烷氧基咔唑:
康铁纯等人建立反相高效液相色谱法(RP-HPLC)测定卡维地洛片有关物质。方法为:采用YMC-Pack Pro C8色谱柱(150 mm× 4.6 mm,5μm),以乙腈-0.02 mol·L-1 磷酸二氢钾溶液(用磷酸调pH 2.0)为流动相进行梯度洗脱,流速1.0 mL·min-1 ,柱温 30℃,检测波长为220 nm(杂质E)和240 nm(其他杂质)。具体实验方法如下:
(1)色谱条件
采用YMC-Pack Pro C8色谱柱(150 mm×4.6 mm, 5μm),以2.72 g·L- 1 磷酸二氢钾溶液(用磷酸调节 pH 2.0)为流动相A,乙腈为流动相B,梯度洗脱(0~ 5 min,20%B;5~20 min,20%B→60%B;20~30 min,60%B;30~30.1 min,60%B→20%B;30.1~35 min,20%B),流速1.0 mL·min-1 ,柱温30℃,检测波长 220 nm(杂质E)和240 nm(其他杂质),进样体积 10μL。
(2)溶液的制备
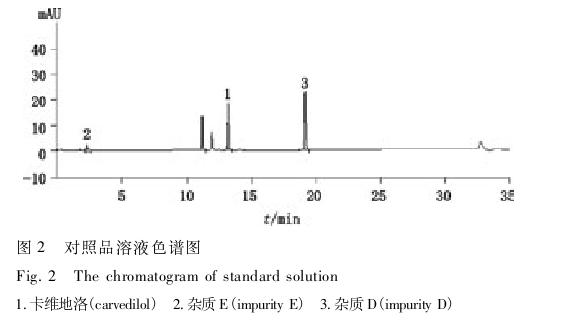
供试品溶液取本品细粉适量(约相当于卡维地洛25 mg),置50 mL量瓶中,加稀释液[流动相A-流动相B(70∶30)]使卡维地洛溶解并稀释至刻度,摇匀,滤过,取续滤液即得。
对照品溶液取杂质D、E和卡维地洛的对照品各适量,精密称定,加稀释液溶解并稀释制成每1 mL 中各约含0.5μg的混合溶液,即得,色谱图见图2。
结果:卡维地洛与已知杂质及强制破坏实验产生的降解产物均能获得良好的分离,杂质D(4-环氧丙烷氧基咔唑)与杂质E[2-(2-甲氧基苯氧基)乙胺]质量浓度分别在0.063 98~ 12.80μg·mL-1 (r=0.999 9)和0.089 33~8.933μg·mL-1 (r=1.000)范围内与峰面积呈良好的线性关系。杂质D高、中、低浓度的回收率(n=3)分别为98.6%、96.5%、94.2%,RSD分别为1.1%、0.8%、1.4%;杂质E高、中、低浓度的回收率(n=3) 分别为99.2%、86%、92.7%,RSD分别为0.15%、0.65%、9.5%。杂质D和杂质E精密度试验的RSD(n=6)分别为1.4%和 0.31%,建立的方法与现行中国药典收载的方法相比能分离检测到更多的杂质。该方法经方法学验证,可用于卡维地洛有关物质的检测和质量控制。
参考文献:
[1]康铁纯,顾宵,何佳佳等.RP-HPLC测定卡维地洛片中的杂质D和杂质E[J].药物分析杂志,2015,35(10):1838-1842.DOI:10.16155/j.0254-1793.2015.10.26.
1
本文将介绍4-羟基-4'-氯二苯甲酮含量测定的方法和步骤,以便准确、可靠地确定4-羟基-4'-氯二苯甲酮的含量。
背景:4-羟基-4'-氯二苯甲酮(4-hydroxy-4'-chlorobenzophenon,简称CBP),分子式为C13H9ClO2 ,分子量为 232.66,为米白色或灰白色至微红白色晶体,熔点为177~181℃,沸点为257℃。4-氯-4’-羟基二苯甲酮(CBP)是合成医药、染料 以及农药等的中间体,尤其广泛用于合成降血脂药物菲诺贝特,但对其纯度的要求较高,即至少 99%,有的甚至要求99.5%以上,否则会影响到后续产品的质量。为保证产品的质量,需建立CBP的含量测定。
1. 合成:
(1)4-羟基-4'-氯二苯甲酮(CBP)的合成
在配有搅拌、回流冷凝器和酸性气体吸收装置的反应烧瓶中,加入27.6 g对羟基苯甲酸(0.2 mol)和200 g氯苯。搅拌升温至60 ℃加入0.1g酰氯化催化剂,30 min内经滴液漏斗加入25 g氯化亚砜(0.21 mol),升温至回流搅拌1 h后冷却至室温。加入66 g无水三氯化铝(0.5 mol),再升温至回流搅拌反应13 h。冷却后将反应液缓慢地倒入500 ml冰水中,剧烈搅拌30 min以分解AlCl3络合物,经分离后水洗有机层至中性,再用水蒸汽蒸馏回收大部分氯苯,将母液冷却至室温,过滤、水洗、干燥得淡棕色粗品37.3 g(红外光谱定性为CBP,熔点:176~179 ℃,HPLC质量分数为95.23%,其主要异构体2-CBP(即4-(2-chlorobenzoyl)phenol)的质量分数为3.93%)。
(2)重结晶提纯
酸碱预处理:将0.2 mol对羟基苯甲酸合成CBP的粗品溶解于200 ml氢氧化钠溶液(wNaOH=4%)中,50 ℃热过滤除去不溶物,滤液用酸析至pH=3,过滤水洗干燥后得白色结晶粉末。
重结晶过程:以粗品2倍重量的异丙醇作溶剂、进行活性炭脱色的重结晶操作得到高纯度CBP的白色结晶(熔点:178.3~179.6℃,HPLC质量分数>99%)。
2. 测定:
林灵超等人立了4-氯-4’-羟基二苯甲酮的高效液相色谱测定方法。以FL2200-Ⅱ为高效液相色谱仪,采用FL-C18为色谱柱,流动相为水(以磷酸调节p H=2.5)∶乙腈=30∶70,检测波长为286 nm,流速为1.0 m L/min,柱温为30℃。所得4-氯-4’-羟基二苯甲酮的线性关系为y=21352x+7 346.8,R2=0.9999,平均回收率为99.42%。该方法简单、准确、重现性好,可用于工业中对4-氯-4’-羟基二苯甲酮的控制。实验方法如下:
(1)标准溶液储备液的配制
精密称取4-氯- 4’-羟基二苯甲酮对照品0.175 3 g,用流动相溶解, 并定容于25 mL白容量瓶中,待用。
(2)标准溶液的配制
移取0.5 mL标准溶液储备液,以流动相稀释并定容于25 m L白容量瓶中,过滤,待用。
(3)样品溶液的配制
精密称取某批次样品 0.074 9 g,用流动相溶解,并定容于25 m L白容量 瓶中,过滤,待用。
(4)色谱条件
色谱柱FL-C18/4.6 mm×250 mm,5μm,流动相水(以磷酸调节pH至2.5)-乙腈=30∶70,流速 1.0 m L/min,柱温30℃,检测波长286 nm,进样量 20μL。
参考文献:
[1]李敢. 4-羟基-4'-氯二苯甲酮的合成研究 [J]. 广州化工, 2014, 42 (11): 27-28.
[2]林灵超. HPLC测定4-氯-4’-羟基二苯甲酮的含量 [J]. 应用化工, 2012, 41 (05): 908-909+913. DOI:10.16581/j.cnki.issn1671-3206.2012.05.034
[3]金宁人,贾建洪,潘志彦. 高纯度4-羟基4’-氯二苯甲酮的合成 [J]. 浙江工业大学学报, 2000, (02): 47-50. 显示全部本文将介绍4-羟基-4'-氯二苯甲酮含量测定的方法和步骤,以便准确、可靠地确定4-羟基-4'-氯二苯甲酮的含量。
背景:4-羟基-4'-氯二苯甲酮(4-hydroxy-4'-chlorobenzophenon,简称CBP),分子式为C13H9ClO2 ,分子量为 232.66,为米白色或灰白色至微红白色晶体,熔点为177~181℃,沸点为257℃。4-氯-4’-羟基二苯甲酮(CBP)是合成医药、染料 以及农药等的中间体,尤其广泛用于合成降血脂药物菲诺贝特,但对其纯度的要求较高,即至少 99%,有的甚至要求99.5%以上,否则会影响到后续产品的质量。为保证产品的质量,需建立CBP的含量测定。
1. 合成:
(1)4-羟基-4'-氯二苯甲酮(CBP)的合成
在配有搅拌、回流冷凝器和酸性气体吸收装置的反应烧瓶中,加入27.6 g对羟基苯甲酸(0.2 mol)和200 g氯苯。搅拌升温至60 ℃加入0.1g酰氯化催化剂,30 min内经滴液漏斗加入25 g氯化亚砜(0.21 mol),升温至回流搅拌1 h后冷却至室温。加入66 g无水三氯化铝(0.5 mol),再升温至回流搅拌反应13 h。冷却后将反应液缓慢地倒入500 ml冰水中,剧烈搅拌30 min以分解AlCl3络合物,经分离后水洗有机层至中性,再用水蒸汽蒸馏回收大部分氯苯,将母液冷却至室温,过滤、水洗、干燥得淡棕色粗品37.3 g(红外光谱定性为CBP,熔点:176~179 ℃,HPLC质量分数为95.23%,其主要异构体2-CBP(即4-(2-chlorobenzoyl)phenol)的质量分数为3.93%)。
(2)重结晶提纯
酸碱预处理:将0.2 mol对羟基苯甲酸合成CBP的粗品溶解于200 ml氢氧化钠溶液(wNaOH=4%)中,50 ℃热过滤除去不溶物,滤液用酸析至pH=3,过滤水洗干燥后得白色结晶粉末。
重结晶过程:以粗品2倍重量的异丙醇作溶剂、进行活性炭脱色的重结晶操作得到高纯度CBP的白色结晶(熔点:178.3~179.6℃,HPLC质量分数>99%)。
2. 测定:
林灵超等人立了4-氯-4’-羟基二苯甲酮的高效液相色谱测定方法。以FL2200-Ⅱ为高效液相色谱仪,采用FL-C18为色谱柱,流动相为水(以磷酸调节p H=2.5)∶乙腈=30∶70,检测波长为286 nm,流速为1.0 m L/min,柱温为30℃。所得4-氯-4’-羟基二苯甲酮的线性关系为y=21352x+7 346.8,R2=0.9999,平均回收率为99.42%。该方法简单、准确、重现性好,可用于工业中对4-氯-4’-羟基二苯甲酮的控制。实验方法如下:
(1)标准溶液储备液的配制
精密称取4-氯- 4’-羟基二苯甲酮对照品0.175 3 g,用流动相溶解, 并定容于25 mL白容量瓶中,待用。
(2)标准溶液的配制
移取0.5 mL标准溶液储备液,以流动相稀释并定容于25 m L白容量瓶中,过滤,待用。
(3)样品溶液的配制
精密称取某批次样品 0.074 9 g,用流动相溶解,并定容于25 m L白容量 瓶中,过滤,待用。
(4)色谱条件
色谱柱FL-C18/4.6 mm×250 mm,5μm,流动相水(以磷酸调节pH至2.5)-乙腈=30∶70,流速 1.0 m L/min,柱温30℃,检测波长286 nm,进样量 20μL。
参考文献:
[1]李敢. 4-羟基-4'-氯二苯甲酮的合成研究 [J]. 广州化工, 2014, 42 (11): 27-28.
[2]林灵超. HPLC测定4-氯-4’-羟基二苯甲酮的含量 [J]. 应用化工, 2012, 41 (05): 908-909+913. DOI:10.16581/j.cnki.issn1671-3206.2012.05.034
[3]金宁人,贾建洪,潘志彦. 高纯度4-羟基4’-氯二苯甲酮的合成 [J]. 浙江工业大学学报, 2000, (02): 47-50.1
本文将介绍如何利用分子印迹技术进行西草净的含量检测,旨在为相关领域的研究人员提供实验支持。
背景:西草净 (simetryn,以下简称SMT) 属三嗪类除草剂,常用于防除稗草、牛毛草等杂草,但不易降解,且具有一定的致癌、致畸、致突变作用。对三嗪类除草剂残留的检测方法主要有气相色谱法、 高效液相色谱法、气相色谱-串联质谱法和液相色谱-串联质谱法。但这些检测技术虽然灵敏度和回收率都能满足检测需求且效果良好,但对溶剂要求严苛,并需要昂贵的仪器和专业分析人员。因此,开发高选择性、高灵敏性的快速检测方法十分必要。
利用分子印迹技术进行含量检测:
分子印迹技术(Molecular Imprinting Technique, MIT)是一种制备对目标分子具有特异识别功能的聚合物的技术,形成的聚合物的内部空腔在尺寸、空间形状和作用点上均与模板分子相匹配,对模板分子具有很高的选择识别性。
1. 西草净分子印迹电化学传感器
陈昱安等人以西草净为模板 分子、甲基丙烯酸为功能单体,采用原位引发聚合法,在玻碳电极表面进行热聚合成膜,制备出西草净分子印迹电化学传感器,并将其用于样品中西草净含量的检测。分子印迹电极的制备具体如下:
(1)聚合溶液的配制
将12.66 mg的SMT 和0.02 mL的MAA放于10 mL乙腈中,超声10 min, 使其完全溶解。然后置于5 ℃的恒温箱中,预聚合12 h。再依次加入0.124 mL的EGDMA和6.25 mg 的AIBN,通氮10 min除氧,得到SMT-MAA聚合溶液。
(2)分子印迹及非分子印迹电极的制备
采用原位引发聚合法制备印迹和非印迹电极。取10 μL 的SMT-MAA聚合溶液,均匀滴涂到GCE表面,置于60 ℃真空干燥箱内热聚合6 h,得到印迹电极。
将该印迹电极放入V(甲醇) : V(乙酸) = 9 : 1混合溶液中,搅拌洗脱12 min,去除模板分子,得到洗脱后的印迹电极,即西草净分子印迹电极 (SMT MIP/GCE),其制备过程及检测原理如图所示。 非印迹电极的制备除不加SMT外,其余步骤与印迹电极的制备相同。
(3)结果
在滴涂量为10 μL、60 ℃下热聚合制备出的西草净电化学传感器 (SMT-MIP/GCE) 具有良好的选择性、重复性和稳定性,其线性范围为0.5~1 μmol/L(1) 和 2~30 μmol/L(2),对应的线性关系分别为I1 = ?3.33c+39.03,I2= ?0.75c+35.52,相关系数分别 为r1 = 0.985,r2 = 0.997,检出限 (LOD) 分别为LOD1 = 0.13 μmol/L和LOD2 = 0.89 μmol/L。将所建立的西草净分子印迹电化学检测方法用于烟叶添加样品提取液中西草净的检测,该印迹电极能够在8 min内完成对烟叶添加样品提取液中西草净的吸附,回收率为76%~88%,相对标准偏差为2.7%~7.6%,该方法能够初步满足烟草中西草净快速检测的需求。
2. 西草净分子印迹光子晶体水凝胶薄膜
光子晶体的三维有序周期结构使其能与可见光发生相互作用,产生布拉格衍射,表现出鲜艳明亮的结构颜色。分子印迹光子晶体水凝胶薄膜(Molecularly Imprinted Photonic Crystal Hydrogels, MIPHs)将这两种技术结合在一起,能对目标分子产生特异识别吸附,吸附目标分子后会产生体积变化导致其晶格参数改变,造成布拉格衍射峰偏移,将分子信号转变为肉眼可观察的光学信号。佟振浩等人以甲基丙烯酸(MAA)为功能单体,乙二醇二甲酸丙烯酸酯(EGDMA) 为交联剂,光引发聚合制备了西草净分子印迹光子晶体水凝胶薄膜。
结果表明,MAA与EGDMA的摩尔比为5:1时,MIPHs的吸附溶胀能力最佳,其在西草 净-乙腈溶液中的最大衍射峰位移达到131nm。将分子印迹-固相萃取(Solid Phase Extraction, SPE)技术与MIPHs相结合,净化和检测样品中的农残。加标烟叶提取液经过固相萃取处理后,活性基质干扰减小了,三嗪类除草剂回收率为83.19-98.62%,MIPHs对其直接进行检测后的布拉格衍射峰位移达到101nm。初步确定MIPHs对实际烟叶提取液的最低检出限为1.0 μg/mL,达到了宏观颜色变化的条件。
参考文献:
[1]佟振浩. 西草净分子印迹光子晶体水凝胶薄膜的制备及其性能研究[D]. 昆明理工大学, 2020. DOI:10.27200/d.cnki.gkmlu.2020.002438
[2]陈昱安,顾丽莉,师君丽等. 西草净分子印迹电化学传感器的制备及应用 [J]. 农药学学报, 2020, 22 (03): 483-492. DOI:10.16801/j.issn.1008-7303.2020.0070
显示全部本文将介绍如何利用分子印迹技术进行西草净的含量检测,旨在为相关领域的研究人员提供实验支持。
背景:西草净 (simetryn,以下简称SMT) 属三嗪类除草剂,常用于防除稗草、牛毛草等杂草,但不易降解,且具有一定的致癌、致畸、致突变作用。对三嗪类除草剂残留的检测方法主要有气相色谱法、 高效液相色谱法、气相色谱-串联质谱法和液相色谱-串联质谱法。但这些检测技术虽然灵敏度和回收率都能满足检测需求且效果良好,但对溶剂要求严苛,并需要昂贵的仪器和专业分析人员。因此,开发高选择性、高灵敏性的快速检测方法十分必要。
利用分子印迹技术进行含量检测:
分子印迹技术(Molecular Imprinting Technique, MIT)是一种制备对目标分子具有特异识别功能的聚合物的技术,形成的聚合物的内部空腔在尺寸、空间形状和作用点上均与模板分子相匹配,对模板分子具有很高的选择识别性。
1. 西草净分子印迹电化学传感器
陈昱安等人以西草净为模板 分子、甲基丙烯酸为功能单体,采用原位引发聚合法,在玻碳电极表面进行热聚合成膜,制备出西草净分子印迹电化学传感器,并将其用于样品中西草净含量的检测。分子印迹电极的制备具体如下:
(1)聚合溶液的配制
将12.66 mg的SMT 和0.02 mL的MAA放于10 mL乙腈中,超声10 min, 使其完全溶解。然后置于5 ℃的恒温箱中,预聚合12 h。再依次加入0.124 mL的EGDMA和6.25 mg 的AIBN,通氮10 min除氧,得到SMT-MAA聚合溶液。
(2)分子印迹及非分子印迹电极的制备
采用原位引发聚合法制备印迹和非印迹电极。取10 μL 的SMT-MAA聚合溶液,均匀滴涂到GCE表面,置于60 ℃真空干燥箱内热聚合6 h,得到印迹电极。
将该印迹电极放入V(甲醇) : V(乙酸) = 9 : 1混合溶液中,搅拌洗脱12 min,去除模板分子,得到洗脱后的印迹电极,即西草净分子印迹电极 (SMT MIP/GCE),其制备过程及检测原理如图所示。 非印迹电极的制备除不加SMT外,其余步骤与印迹电极的制备相同。
(3)结果
在滴涂量为10 μL、60 ℃下热聚合制备出的西草净电化学传感器 (SMT-MIP/GCE) 具有良好的选择性、重复性和稳定性,其线性范围为0.5~1 μmol/L(1) 和 2~30 μmol/L(2),对应的线性关系分别为I1 = ?3.33c+39.03,I2= ?0.75c+35.52,相关系数分别 为r1 = 0.985,r2 = 0.997,检出限 (LOD) 分别为LOD1 = 0.13 μmol/L和LOD2 = 0.89 μmol/L。将所建立的西草净分子印迹电化学检测方法用于烟叶添加样品提取液中西草净的检测,该印迹电极能够在8 min内完成对烟叶添加样品提取液中西草净的吸附,回收率为76%~88%,相对标准偏差为2.7%~7.6%,该方法能够初步满足烟草中西草净快速检测的需求。
2. 西草净分子印迹光子晶体水凝胶薄膜
光子晶体的三维有序周期结构使其能与可见光发生相互作用,产生布拉格衍射,表现出鲜艳明亮的结构颜色。分子印迹光子晶体水凝胶薄膜(Molecularly Imprinted Photonic Crystal Hydrogels, MIPHs)将这两种技术结合在一起,能对目标分子产生特异识别吸附,吸附目标分子后会产生体积变化导致其晶格参数改变,造成布拉格衍射峰偏移,将分子信号转变为肉眼可观察的光学信号。佟振浩等人以甲基丙烯酸(MAA)为功能单体,乙二醇二甲酸丙烯酸酯(EGDMA) 为交联剂,光引发聚合制备了西草净分子印迹光子晶体水凝胶薄膜。
结果表明,MAA与EGDMA的摩尔比为5:1时,MIPHs的吸附溶胀能力最佳,其在西草 净-乙腈溶液中的最大衍射峰位移达到131nm。将分子印迹-固相萃取(Solid Phase Extraction, SPE)技术与MIPHs相结合,净化和检测样品中的农残。加标烟叶提取液经过固相萃取处理后,活性基质干扰减小了,三嗪类除草剂回收率为83.19-98.62%,MIPHs对其直接进行检测后的布拉格衍射峰位移达到101nm。初步确定MIPHs对实际烟叶提取液的最低检出限为1.0 μg/mL,达到了宏观颜色变化的条件。
参考文献:
[1]佟振浩. 西草净分子印迹光子晶体水凝胶薄膜的制备及其性能研究[D]. 昆明理工大学, 2020. DOI:10.27200/d.cnki.gkmlu.2020.002438
[2]陈昱安,顾丽莉,师君丽等. 西草净分子印迹电化学传感器的制备及应用 [J]. 农药学学报, 2020, 22 (03): 483-492. DOI:10.16801/j.issn.1008-7303.2020.0070
1
本文将介绍测定4-氯-3-磺酰胺基苯甲酸中氯磺酸残留量的方法,以期为分析化学和医药等领域的相关研究人员提供实验支持。
简述:4-氯-3-磺酰胺基苯甲酸,英文名称:4-Chloro-5-sulphamoylbenzoic acid,CAS:1205-30-7,分子式:C7H6ClNO4S,外观与性状:白色粉末。4-氯-3-磺酰胺基苯甲酸常用于药物布美他尼的合成。
测定4-氯-3-磺酰胺基苯甲酸中氯磺酸残留量:
吲达帕胺是由法国 Servier 公司研制的长效抗高血压药,具有较强的血管扩张作用和一定的利尿效果。针对吲达帕胺作为常用抗高血压药的特性,对其原料药中潜在的基因毒性杂质进行全面研究具有重要的实际意义。吲达帕胺的最大日服用剂量(MDD)为10mg/天,因此根据TTC计算,原料药中基因毒性杂质的可接受浓度为150ppm。4-氯-3-磺酰胺基苯甲酸是吲达帕胺生产工艺的关键起始物料,其中潜在的基因毒性杂质可能会带入吲达帕胺中。氯磺酸作为潜在的基因毒性杂质,根据法规要求,必须对其残留量进行检测和控制。即氯磺酸在最终 API中的含量报告值应低于由 TCC 推算出的接受标准 150ppm。
氯磺酸是一种强氧化剂,与水接触会剧烈分解,释放大量热量和浓烟。在潮湿空气中与金属接触时,会腐蚀金属并释放氢气,易于引发燃烧爆炸。当与易燃物(如苯)和可燃物(如糖、纤维素等)接触时,会发生剧烈反应,甚至引发燃烧,同时具有强腐蚀性。因此,一般的分析方法无法对其残留量进行有效检测。谢玲玲等人开发了离子色谱法对其进行监控,并为了保证实验的安全性和有效性,利用氯磺酸遇水即产生水解反应的特点(反应方程式:HSO3Cl + H2O = H2SO4 + HCl),采用无水硫酸钠替代氯磺酸配制对照溶液,即 1g 氯磺酸相当于 1.2g 硫酸钠。分析方法具体如下:
1. 色谱条件
检测器:ECD,流速:1.0ml/min,柱温:室温(25℃),进样量:20μl,运行时间:20min。
2. 溶液配制
2.1 淋洗液:称取碳酸氢钠 0.24g,碳酸钠 0.95g,加蒸馏水 2L 使溶解,用 0.45μm 的滤膜过滤。
2.2 再生液:取硫酸 2mL 加入到 2L 蒸馏水中,混匀,用 0.45μm 的滤膜过滤。
2.3 硫酸钠储备液:
(1)溶液①:称取无水硫酸钠 54mg 用蒸馏水稀释至 100.0mL。
(2)溶液②(即“硫酸钠储备液”):取溶液①溶液 2.0mL,用蒸馏水稀释至 100.0mL。
2.4 对照溶液:取“硫酸钠储备液”5.0mL,用蒸馏水稀释至 50.0mL。
2.5 样品溶液:称取试样 400mg 加蒸馏水至 20.0mL,超声震荡 10min,用0.22μm 过滤膜过滤后使用。
2.6 专属性溶液:称取试样 400mg,加入“硫酸钠储备液”2.0mL,加蒸馏水至 20.0mL,超声震荡 10min,用 0.22μm 过滤膜过滤后使用。
2.7 精密度溶液:称取试样 400mg,加入“硫酸钠储备液”4.0mL,加蒸馏水至 20.0mL,超声震荡 10min,用 0.22μm 过滤膜过滤后使用。(将此溶液重复配制 6 份,作为精密度溶液①~⑥)
2.8 定量限(LOQ)溶液:取对照溶液逐步稀释。
2.9 检测线(LOD)溶液:取定量限(LOQ)溶液逐步稀释。
3. 结果
该方法适用于 4-氯-3-磺酰胺基苯甲酸中氯磺酸残留的检测,检出限为11.22μg/ml,定量限为 44.88μg/ml。6 批样品的检测结果均小于 45ppm,根据EMA 发布的 EMA/CHMP/SWP/431994/2007《基因毒性杂质限度指南问答》,氯磺酸在 6 批 4-氯-3-磺酰胺基苯甲酸中残留量都小于 45ppm(可接受浓度150ppm 的 30%),故氯磺酸无需在原料药吲达帕胺中进行考察。
参考文献:
[1] 谢玲玲,李贺然. 离子色谱法测定4-氯-3-磺酰胺基苯甲酸中氯磺酸残留量[J]. 中国化工贸易,2015,7(33):11-12. DOI:10.3969/j.issn.1674-5167.2015.33.009.
显示全部本文将介绍测定4-氯-3-磺酰胺基苯甲酸中氯磺酸残留量的方法,以期为分析化学和医药等领域的相关研究人员提供实验支持。
简述:4-氯-3-磺酰胺基苯甲酸,英文名称:4-Chloro-5-sulphamoylbenzoic acid,CAS:1205-30-7,分子式:C7H6ClNO4S,外观与性状:白色粉末。4-氯-3-磺酰胺基苯甲酸常用于药物布美他尼的合成。
测定4-氯-3-磺酰胺基苯甲酸中氯磺酸残留量:
吲达帕胺是由法国 Servier 公司研制的长效抗高血压药,具有较强的血管扩张作用和一定的利尿效果。针对吲达帕胺作为常用抗高血压药的特性,对其原料药中潜在的基因毒性杂质进行全面研究具有重要的实际意义。吲达帕胺的最大日服用剂量(MDD)为10mg/天,因此根据TTC计算,原料药中基因毒性杂质的可接受浓度为150ppm。4-氯-3-磺酰胺基苯甲酸是吲达帕胺生产工艺的关键起始物料,其中潜在的基因毒性杂质可能会带入吲达帕胺中。氯磺酸作为潜在的基因毒性杂质,根据法规要求,必须对其残留量进行检测和控制。即氯磺酸在最终 API中的含量报告值应低于由 TCC 推算出的接受标准 150ppm。
氯磺酸是一种强氧化剂,与水接触会剧烈分解,释放大量热量和浓烟。在潮湿空气中与金属接触时,会腐蚀金属并释放氢气,易于引发燃烧爆炸。当与易燃物(如苯)和可燃物(如糖、纤维素等)接触时,会发生剧烈反应,甚至引发燃烧,同时具有强腐蚀性。因此,一般的分析方法无法对其残留量进行有效检测。谢玲玲等人开发了离子色谱法对其进行监控,并为了保证实验的安全性和有效性,利用氯磺酸遇水即产生水解反应的特点(反应方程式:HSO3Cl + H2O = H2SO4 + HCl),采用无水硫酸钠替代氯磺酸配制对照溶液,即 1g 氯磺酸相当于 1.2g 硫酸钠。分析方法具体如下:
1. 色谱条件
检测器:ECD,流速:1.0ml/min,柱温:室温(25℃),进样量:20μl,运行时间:20min。
2. 溶液配制
2.1 淋洗液:称取碳酸氢钠 0.24g,碳酸钠 0.95g,加蒸馏水 2L 使溶解,用 0.45μm 的滤膜过滤。
2.2 再生液:取硫酸 2mL 加入到 2L 蒸馏水中,混匀,用 0.45μm 的滤膜过滤。
2.3 硫酸钠储备液:
(1)溶液①:称取无水硫酸钠 54mg 用蒸馏水稀释至 100.0mL。
(2)溶液②(即“硫酸钠储备液”):取溶液①溶液 2.0mL,用蒸馏水稀释至 100.0mL。
2.4 对照溶液:取“硫酸钠储备液”5.0mL,用蒸馏水稀释至 50.0mL。
2.5 样品溶液:称取试样 400mg 加蒸馏水至 20.0mL,超声震荡 10min,用0.22μm 过滤膜过滤后使用。
2.6 专属性溶液:称取试样 400mg,加入“硫酸钠储备液”2.0mL,加蒸馏水至 20.0mL,超声震荡 10min,用 0.22μm 过滤膜过滤后使用。
2.7 精密度溶液:称取试样 400mg,加入“硫酸钠储备液”4.0mL,加蒸馏水至 20.0mL,超声震荡 10min,用 0.22μm 过滤膜过滤后使用。(将此溶液重复配制 6 份,作为精密度溶液①~⑥)
2.8 定量限(LOQ)溶液:取对照溶液逐步稀释。
2.9 检测线(LOD)溶液:取定量限(LOQ)溶液逐步稀释。
3. 结果
该方法适用于 4-氯-3-磺酰胺基苯甲酸中氯磺酸残留的检测,检出限为11.22μg/ml,定量限为 44.88μg/ml。6 批样品的检测结果均小于 45ppm,根据EMA 发布的 EMA/CHMP/SWP/431994/2007《基因毒性杂质限度指南问答》,氯磺酸在 6 批 4-氯-3-磺酰胺基苯甲酸中残留量都小于 45ppm(可接受浓度150ppm 的 30%),故氯磺酸无需在原料药吲达帕胺中进行考察。
参考文献:
[1] 谢玲玲,李贺然. 离子色谱法测定4-氯-3-磺酰胺基苯甲酸中氯磺酸残留量[J]. 中国化工贸易,2015,7(33):11-12. DOI:10.3969/j.issn.1674-5167.2015.33.009.
1
本文将介绍关于4-氯水杨酸的太赫兹光谱弱相互作用分析,旨在为相关研究人员提供参考依据和实验支持。
简述:4-氯水杨酸,英文名称:4-Chlorosalicylic acid,CAS:5106-98-9,分子式:C7H5ClO3,外观与性状:灰白色至淡米色粉末。4-氯水杨酸是除草剂Benthiocarb的代谢产物。4-氯水杨酸具有抗真菌活性。
晶体中分子间存在的氢键、卤键、范德华力以及π···π相互作用等弱相互作用均落在太赫兹波段,这些作用对于晶体的结构、对称性及其稳定性等具有重要的影响,目前已广泛应用于化学、生物、物理、医学等众多领域,而且与许多生命现象密切相关。水杨酸及其衍生物分子由于本身带有羰基供电子基团和羟基缺电子基团,因而在形成的晶体中也可能存在多种组合的分子间弱相互作用。
4-氯水杨酸的太赫兹实验与理论研究:
Hirshfeld 表面分析是一种研究分子晶体间弱相互作用极为有用的方法,它以一种新颖的视觉方式反映了分子间的相互作用,能够非常清晰地描绘出分子在结晶环境中的未知形状,并从分子间相互作用的界面不同区域的颜色上,能立刻了解到什么位置发生了什么类型的作用。并且还能绘制指纹图来定量研究氢键、卤键、范德华相互作用,C-H…π相互作用,π…π堆积等分子间弱相互作用,因而更加便于人们进行识别和判断。
贺微等人运用THz-TDS 技术对水杨酸的两个衍生物4-氯水杨酸和5-氯水杨酸进行了实验测量,并利用量子化学理论中密度泛函理论对它们的单分子和分子团簇构型分别进行了理论模拟计算, 通过PED分析每种基团对简正振动模式贡献的百分比,并利用Hirshfeld表面分析的 Dorm表面图和指纹图对4-氯水杨酸和5-氯水杨酸晶体中的分子间弱相互作用的位置和类型进行了可视化。
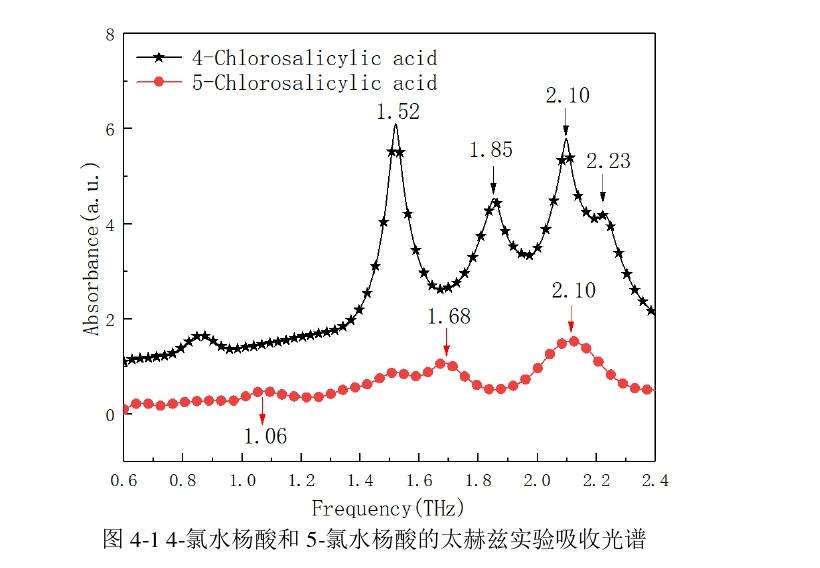
(1)太赫兹实验光谱
水杨酸的两种衍生物在室温下测得的吸收光谱如图4-1所示,可以看出,4-氯水杨酸一共出现了四个较窄特征吸收峰,强吸收峰出现在1.52、1.85和2.10 THz,而在 2.23 THz出现弱的吸收峰。
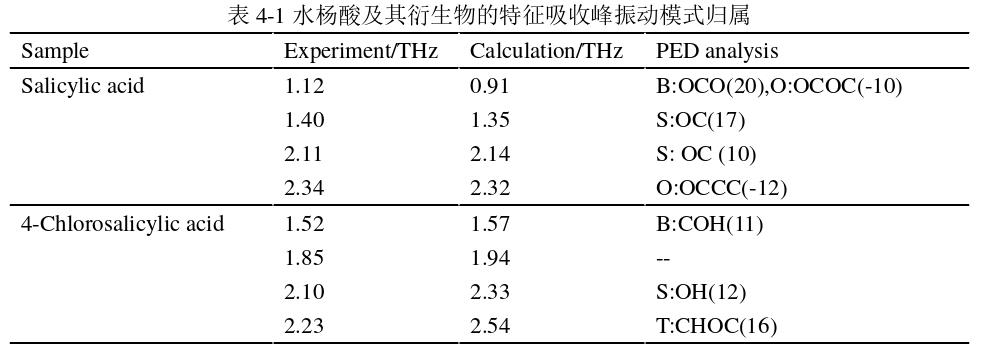
(2)4-氯水杨酸的特征吸收峰振动模式归属
从表4-1可以看出,4-氯水杨酸分子团簇在1.57THz处的振动模式主要是C129O107H218原子所属分子的键角弯曲;1.94THz处的振动模式特征未能够被指认出来,是因为PED 分析中只有贡献大于5%的才能被输出;2.33THz处的振动方式主要为O77H99原子所在分子的键长伸缩;2.54THz处的振动方式C176H182O160C186原子所属基团的二面角扭转。4-氯水杨酸分子团簇的特征振动模式存在键角弯曲、键长伸缩和二面体扭转三种振动。

(3)Hirshfeld表面分析4-氯水杨酸分子晶体的弱相互作用
为了更直观地观察到水杨酸的衍生物在哪里以及发生了什么类型的弱相互作用,使用multiwfn结合VMD程序绘制水杨酸晶体结构的Hirshfeld 表面图,通过 Hirshfeld表面分析4-氯水杨酸和5-氯水杨酸晶体之间的分子间相互作用。红色区域 代表电子密度大,这是由于形成了氢键。蓝色区域代表电子密度很小,没有明显的相互作用。白色区域代表电子密度中等,对应稍弱相互作用。
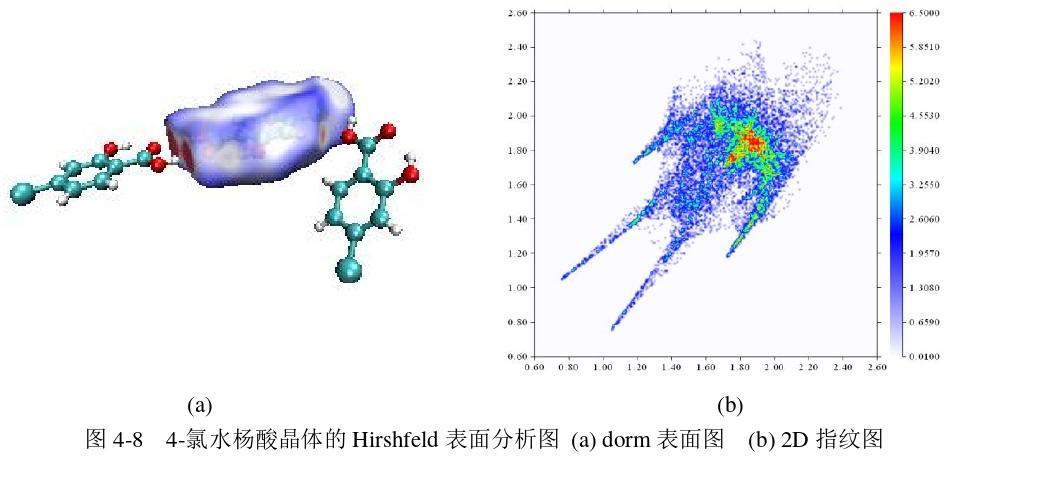
图4-8(a)是4-氯水杨酸的Hirshfeld 表面分析的dorm表面图。图4-8(a)dnorm表面图左边出现了两个深红色区域,是中心分子上的羰基O与羟基H的相互作用,右边出现了一个淡红色区域,是周围分子上的羰基O与中心分子H相互作用形成。从图4-8(b)水杨酸2D指纹图中可以看出,左下两个尖刺之间弥散的点是典型的 H···H间的相互作用。Hirshfeld表面贡献最大的是 H···Cl 间的相互作用,在指纹图上面出现的两个弥散的点出现组成的蓝色翅膀,就是C-H···Cl接触的点。左下方有一对尖刺,这是中心分子上羰基O 与羟基H形成的氢键,上面的一个尖刺对应于氢键供体,而下面的一个尖刺对应于氢键受体。在(1.65,1.65)和(1.85,1.85)之间,有一部分蓝绿色区域,这个区域的点分布 非常窄,它与氢键尖刺非常类似,但含有许多橙色和红色的点,这是因为 Cl···Cl 之 间形成了卤键。此外,在指纹图中对角线上 1.8?附近的蓝绿色区域是典型的π···π堆积作用。
参考文献:
[1]贺微. 水杨酸及其衍生物的太赫兹光谱弱相互作用分析[D]. 桂林电子科技大学, 2021. DOI:10.27049/d.cnki.ggldc.2021.000319.
[2]龚润军,周宇涵,苗蔚荣等. 2-溴-6-氯-3-(2H)-苯并呋喃酮的合成 [J]. 精细与专用化学品, 2005, (07): 12-13+17. 显示全部本文将介绍关于4-氯水杨酸的太赫兹光谱弱相互作用分析,旨在为相关研究人员提供参考依据和实验支持。
简述:4-氯水杨酸,英文名称:4-Chlorosalicylic acid,CAS:5106-98-9,分子式:C7H5ClO3,外观与性状:灰白色至淡米色粉末。4-氯水杨酸是除草剂Benthiocarb的代谢产物。4-氯水杨酸具有抗真菌活性。
晶体中分子间存在的氢键、卤键、范德华力以及π···π相互作用等弱相互作用均落在太赫兹波段,这些作用对于晶体的结构、对称性及其稳定性等具有重要的影响,目前已广泛应用于化学、生物、物理、医学等众多领域,而且与许多生命现象密切相关。水杨酸及其衍生物分子由于本身带有羰基供电子基团和羟基缺电子基团,因而在形成的晶体中也可能存在多种组合的分子间弱相互作用。
4-氯水杨酸的太赫兹实验与理论研究:
Hirshfeld 表面分析是一种研究分子晶体间弱相互作用极为有用的方法,它以一种新颖的视觉方式反映了分子间的相互作用,能够非常清晰地描绘出分子在结晶环境中的未知形状,并从分子间相互作用的界面不同区域的颜色上,能立刻了解到什么位置发生了什么类型的作用。并且还能绘制指纹图来定量研究氢键、卤键、范德华相互作用,C-H…π相互作用,π…π堆积等分子间弱相互作用,因而更加便于人们进行识别和判断。
贺微等人运用THz-TDS 技术对水杨酸的两个衍生物4-氯水杨酸和5-氯水杨酸进行了实验测量,并利用量子化学理论中密度泛函理论对它们的单分子和分子团簇构型分别进行了理论模拟计算, 通过PED分析每种基团对简正振动模式贡献的百分比,并利用Hirshfeld表面分析的 Dorm表面图和指纹图对4-氯水杨酸和5-氯水杨酸晶体中的分子间弱相互作用的位置和类型进行了可视化。
(1)太赫兹实验光谱
水杨酸的两种衍生物在室温下测得的吸收光谱如图4-1所示,可以看出,4-氯水杨酸一共出现了四个较窄特征吸收峰,强吸收峰出现在1.52、1.85和2.10 THz,而在 2.23 THz出现弱的吸收峰。
(2)4-氯水杨酸的特征吸收峰振动模式归属
从表4-1可以看出,4-氯水杨酸分子团簇在1.57THz处的振动模式主要是C129O107H218原子所属分子的键角弯曲;1.94THz处的振动模式特征未能够被指认出来,是因为PED 分析中只有贡献大于5%的才能被输出;2.33THz处的振动方式主要为O77H99原子所在分子的键长伸缩;2.54THz处的振动方式C176H182O160C186原子所属基团的二面角扭转。4-氯水杨酸分子团簇的特征振动模式存在键角弯曲、键长伸缩和二面体扭转三种振动。
(3)Hirshfeld表面分析4-氯水杨酸分子晶体的弱相互作用
为了更直观地观察到水杨酸的衍生物在哪里以及发生了什么类型的弱相互作用,使用multiwfn结合VMD程序绘制水杨酸晶体结构的Hirshfeld 表面图,通过 Hirshfeld表面分析4-氯水杨酸和5-氯水杨酸晶体之间的分子间相互作用。红色区域 代表电子密度大,这是由于形成了氢键。蓝色区域代表电子密度很小,没有明显的相互作用。白色区域代表电子密度中等,对应稍弱相互作用。
图4-8(a)是4-氯水杨酸的Hirshfeld 表面分析的dorm表面图。图4-8(a)dnorm表面图左边出现了两个深红色区域,是中心分子上的羰基O与羟基H的相互作用,右边出现了一个淡红色区域,是周围分子上的羰基O与中心分子H相互作用形成。从图4-8(b)水杨酸2D指纹图中可以看出,左下两个尖刺之间弥散的点是典型的 H···H间的相互作用。Hirshfeld表面贡献最大的是 H···Cl 间的相互作用,在指纹图上面出现的两个弥散的点出现组成的蓝色翅膀,就是C-H···Cl接触的点。左下方有一对尖刺,这是中心分子上羰基O 与羟基H形成的氢键,上面的一个尖刺对应于氢键供体,而下面的一个尖刺对应于氢键受体。在(1.65,1.65)和(1.85,1.85)之间,有一部分蓝绿色区域,这个区域的点分布 非常窄,它与氢键尖刺非常类似,但含有许多橙色和红色的点,这是因为 Cl···Cl 之 间形成了卤键。此外,在指纹图中对角线上 1.8?附近的蓝绿色区域是典型的π···π堆积作用。
参考文献:
[1]贺微. 水杨酸及其衍生物的太赫兹光谱弱相互作用分析[D]. 桂林电子科技大学, 2021. DOI:10.27049/d.cnki.ggldc.2021.000319.
[2]龚润军,周宇涵,苗蔚荣等. 2-溴-6-氯-3-(2H)-苯并呋喃酮的合成 [J]. 精细与专用化学品, 2005, (07): 12-13+17.1
随着1,3-二氯-5,5-二甲基海因应用范围的扩大,对其含量进行准确测定变得至关重要。
背景:1,3-二氯-5,5-二甲基海因,又名1,3-二氯-5,5-二甲基乙内酰脲,简称二氯海因或DCDMH,是一种新型的氯(亚)胺型消毒杀菌剂和漂白剂,可广泛应用于游泳池消毒、饮用水消毒、水产养殖、工业循环水处理、餐具消毒、食品与化妆品生产设备消毒及环境消毒等领域。与目前消毒剂行业产量最大的氯胺型消毒杀菌剂氯代异氰尿酸相比,它具有气味小、毒性小、贮存稳定性好、水解残留物降解快等优点。
但是,其作为定时释放有效成分(有效氯)的杀菌剂及消毒剂,其消毒能力及杀菌能力是随时间变化的,因此需要随时检测其有效成分的浓度,以便对其在水中的杀菌能力及消毒能力作出准确判断。
1. 合成:
在装有搅拌器的二颈烧瓶中,加入12.8 g(0.1 mol)5,5-二甲基海因、80 mL水和8.4 g氢氧化钠,搅拌溶解、冰水浴冷至5~6 ℃,在剧烈搅拌下通氯气,至反应体系为浅黄绿色。抽滤、水洗(30 mL×3),晾干,得1,3-二氯-5,5-二甲基海因18.9 g,收率96.0%,m.p.129~130 ℃。
2. 测定:
饶志明等人研究了在碱性条件下 ( pH:12.0— 12.5) 1,3-二氯 - 5,5-二甲基海因与鲁米诺 -过氧化氢体系产生的化学发光性质 ,首次建立了一种测定其含量的新方法 ,并应用于游泳池水中 1,3-二氯 - 5,5-二甲基海因的测定 ,结果满意。1,3-二氯 - 5,5-二甲基海因的浓度在 8.0× 10-8— 5.0×10 -6mol/ L范围内 ,与化学发光强度呈良好的线性关系。方法的线性范围宽 ,测定 5.0× 10 -7mol/ L的 1,3-二氯-5,5-二甲基海因 11次 ,求得相对标准偏差为 4.7% ,回收率为 83%—90 % ,方法的检出限为 5.0× 10-8mol/ L,灵敏度高。实验方法为:
(1)1,3-二氯-5,5-二甲基海因标准溶液:取0.1970g的标准样品(含量>99%),加入15mL丙酮溶解,后加水定容为1000 mL,配成1mmol/L的标准溶液,使用时用水稀释至需要的浓度;鲁米诺溶液:储备液浓度为2.0×10-2mol/L(在0.1mol/L氢氧化钠溶液中),使用时稀释为2.0×10-4mol/L;过氧化氢溶液:8.0×10-2mol/L,使用时用水逐级稀释;其他试剂均为分析纯,水为二次去离子水。
(2)实验方法:实验装置如图所示,将一定体积的1,3-二氯-5,5-二甲基海因的标准溶液或样品溶液注入到鲁米诺与过氧化氢混合液的载流中,记录发光信号,以峰高定量。

参考文献:
[1]许延峰,夏永,张选民. 1,3-二氯-5,5-二甲基海因的合成研究 [J]. 氯碱工业, 2002, (06): 34-36.
[2]饶志明,张新荣. 化学发光法直接测定水中1,3-二氯-5,5-二甲基海因的研究 [J]. 光谱实验室, 2001, (03): 294-297.
[3]胡艾希,周宏伟,文耀智等. 1,3-二氯-5,5-二甲基海因 显示全部随着1,3-二氯-5,5-二甲基海因应用范围的扩大,对其含量进行准确测定变得至关重要。
背景:1,3-二氯-5,5-二甲基海因,又名1,3-二氯-5,5-二甲基乙内酰脲,简称二氯海因或DCDMH,是一种新型的氯(亚)胺型消毒杀菌剂和漂白剂,可广泛应用于游泳池消毒、饮用水消毒、水产养殖、工业循环水处理、餐具消毒、食品与化妆品生产设备消毒及环境消毒等领域。与目前消毒剂行业产量最大的氯胺型消毒杀菌剂氯代异氰尿酸相比,它具有气味小、毒性小、贮存稳定性好、水解残留物降解快等优点。
但是,其作为定时释放有效成分(有效氯)的杀菌剂及消毒剂,其消毒能力及杀菌能力是随时间变化的,因此需要随时检测其有效成分的浓度,以便对其在水中的杀菌能力及消毒能力作出准确判断。
1. 合成:
在装有搅拌器的二颈烧瓶中,加入12.8 g(0.1 mol)5,5-二甲基海因、80 mL水和8.4 g氢氧化钠,搅拌溶解、冰水浴冷至5~6 ℃,在剧烈搅拌下通氯气,至反应体系为浅黄绿色。抽滤、水洗(30 mL×3),晾干,得1,3-二氯-5,5-二甲基海因18.9 g,收率96.0%,m.p.129~130 ℃。
2. 测定:
饶志明等人研究了在碱性条件下 ( pH:12.0— 12.5) 1,3-二氯 - 5,5-二甲基海因与鲁米诺 -过氧化氢体系产生的化学发光性质 ,首次建立了一种测定其含量的新方法 ,并应用于游泳池水中 1,3-二氯 - 5,5-二甲基海因的测定 ,结果满意。1,3-二氯 - 5,5-二甲基海因的浓度在 8.0× 10-8— 5.0×10 -6mol/ L范围内 ,与化学发光强度呈良好的线性关系。方法的线性范围宽 ,测定 5.0× 10 -7mol/ L的 1,3-二氯-5,5-二甲基海因 11次 ,求得相对标准偏差为 4.7% ,回收率为 83%—90 % ,方法的检出限为 5.0× 10-8mol/ L,灵敏度高。实验方法为:
(1)1,3-二氯-5,5-二甲基海因标准溶液:取0.1970g的标准样品(含量>99%),加入15mL丙酮溶解,后加水定容为1000 mL,配成1mmol/L的标准溶液,使用时用水稀释至需要的浓度;鲁米诺溶液:储备液浓度为2.0×10-2mol/L(在0.1mol/L氢氧化钠溶液中),使用时稀释为2.0×10-4mol/L;过氧化氢溶液:8.0×10-2mol/L,使用时用水逐级稀释;其他试剂均为分析纯,水为二次去离子水。
(2)实验方法:实验装置如图所示,将一定体积的1,3-二氯-5,5-二甲基海因的标准溶液或样品溶液注入到鲁米诺与过氧化氢混合液的载流中,记录发光信号,以峰高定量。
参考文献:
[1]许延峰,夏永,张选民. 1,3-二氯-5,5-二甲基海因的合成研究 [J]. 氯碱工业, 2002, (06): 34-36.
[2]饶志明,张新荣. 化学发光法直接测定水中1,3-二氯-5,5-二甲基海因的研究 [J]. 光谱实验室, 2001, (03): 294-297.
[3]胡艾希,周宏伟,文耀智等. 1,3-二氯-5,5-二甲基海因1
本文将探讨如何测定吉非替尼中相关杂质4-氯-3-氟苯胺,旨在为相关领域的研究人员提供参考思路和启示。
简述:4-氯-3-氟苯胺,英文名称:4-Chloro-3-fluoroaniline,CAS:367-22-6,分子式:C6H5ClFN。4-氯-3-氟苯胺用于合成甲基5-氯-4-氟-1H-吲哚-2-羧酸甲酯,这是HIV非核苷逆转录酶的磷酸吲哚抑制剂的关键中间体。4-氯-3-氟苯胺还用于合成3-去氯-4-去氟-4-氯-3-氟吉非替尼,这是酪氨酸激酶抑制剂吉非替尼的杂质。
测定:
吉非替尼是一种选择性表皮生长因子受体酪氨酸激酶抑制剂,用于治疗铂剂化疗和多烯紫杉醇化疗均失败的局部晚期或转移性非小细胞(型)肺癌,是首个应用于肺癌治疗的靶向药物。该药物的合成主要原料是3-氯-4-氟苯胺,而在合成3-氯-4-氟苯胺的过程中产生3,4-二氟苯胺和4-氯-3-氟苯胺作为附属产物。由于这些附属产物具有基因毒性,因此在吉非替尼的生产过程中需要控制这些基因毒性杂质的含量。
王珊珊等人建立了液相色谱-串联质谱法测定吉非替尼中基因毒性杂质的含量。方法具体为:采用Accucore C18色谱柱(2.1 mm×100 mm,2.6μm),以0.1%甲酸水溶液-0.1%甲酸乙腈溶液(70:30)为流动相,流速0.3 mL·min-1,柱温30℃;采用ESI离子源正离子模式,多反应离子监测(MRM)模式下选择离子对m/z130.1→83.1(3,4-二氟苯胺)和m/z146.0→111.0(4-氯-3-氟苯胺)进行测定。
基因毒性杂质3,4-二氟苯胺和4-氯-3-氟苯胺的线性浓度范围为124 ng·mL-1,且线性关系良好(r=0.999 4、r=0.999 2);检测限为0.2 mg·kg-1,定量限为0.5 mg·kg-1;3,4-二氟苯胺和4-氯-3-氟苯胺的精密度试验的RSD(n=6)分别为2.3%和2.0%;杂质3,4-二氟苯胺低、中、高浓度的加标回收率(n=3)分别为99.4%、97.0%、105.5%,RSD=8.2%、5.1%、1.7%;杂质4-氯-3-氟苯胺低、中、高浓度的加标回收率(n=3)分别为102.0%、98.6%、106.0%,RSD分别为3.9%、7.9%、2.4%。经检测,3批吉非替尼供试品中3,4-二氟苯胺均未检出,批号2吉非替尼样品中4-氯-3-氟苯胺有少量检出,但低于样品的定量限(<0.5 mg·kg-1)。该方法操作简便,结果可靠,经方法学验证,可用于吉非替尼药物中基因毒性杂质3,4-二氟苯胺和4-氯-3-氟苯胺含量的同时测定。实验方法具体为:
(1)色谱条件:
采用色谱柱Accucore C18(2.1 mm× 100 mm,2.6 μm;填料:十八烷基硅烷键合硅胶),以0.1%甲酸水溶液-0.1%甲酸乙腈溶液(70∶30)为流动相,流速0.3 mL·min-1,柱温30 ℃,进样体积2 μL。
(2)质谱条件:
采用ESI离子源正离子模式,多反应离子监测(MRM)模式下选择离子对m/z 130.1→83.1(3,4-二氟苯胺)和m/z 146.0→111.0(4-氯-3-氟苯胺);解簇电压90 V(3,4-二氟苯胺)、76 V(4- 氯-3-氟苯胺);解离能量32 V(3,4-二氟苯胺)、26 V(4-氯-3-氟苯胺);入口电压10 V;出口电压 12 V;碰撞气压力Medium;气帘气压力1.03 MPa;雾化压力0.38 MPa;辅助加热气压力0.38 MPa;离子喷雾电压5.5 kV;离子源温度500 ℃。
(3)对照品溶液:
精密称取3,4-二氟苯胺、4- 氯-3-氟苯胺对照品各约10.01 mg,置100 mL量瓶中,加稀释剂乙腈-水(50:50)溶解并稀释至刻度,摇匀;精密量取1 mL,置100 mL量瓶中,用稀释剂乙腈-水(50:50)稀释至刻度,摇匀,配制成浓度为 1 μg·mL-1对照品储备液;精密量取1 mL,置10 mL 量瓶中,用稀释剂乙腈-水(50:50)稀释至刻度,摇匀;精密量取12 mL,置100 mL量瓶中,用稀释剂乙腈-水(50:50)稀释至刻度,摇匀,配成浓度为12 ng·mL-1的对照品溶液。
(4)供试品溶液 精密称取吉非替尼供试品约
20.03 mg,置10 mL量瓶中,用稀释剂乙腈-水(50:50) 振摇使溶解并稀释至刻度,取1 mL置2 mL离心管中,12 000 r·min-1离心10 min后取上清液,配制成浓度为2 mg·mL-1的吉非替尼供试品溶液。
参考文献:
[1]王珊珊,宁凡盛,王晓利等.HPLC-MS/MS法分析吉非替尼中痕量基因毒性杂质[J].药物分析杂志,2017,37(07):1309-1313.DOI:10.16155/j.0254-1793.2017.07.23
显示全部本文将探讨如何测定吉非替尼中相关杂质4-氯-3-氟苯胺,旨在为相关领域的研究人员提供参考思路和启示。
简述:4-氯-3-氟苯胺,英文名称:4-Chloro-3-fluoroaniline,CAS:367-22-6,分子式:C6H5ClFN。4-氯-3-氟苯胺用于合成甲基5-氯-4-氟-1H-吲哚-2-羧酸甲酯,这是HIV非核苷逆转录酶的磷酸吲哚抑制剂的关键中间体。4-氯-3-氟苯胺还用于合成3-去氯-4-去氟-4-氯-3-氟吉非替尼,这是酪氨酸激酶抑制剂吉非替尼的杂质。
测定:
吉非替尼是一种选择性表皮生长因子受体酪氨酸激酶抑制剂,用于治疗铂剂化疗和多烯紫杉醇化疗均失败的局部晚期或转移性非小细胞(型)肺癌,是首个应用于肺癌治疗的靶向药物。该药物的合成主要原料是3-氯-4-氟苯胺,而在合成3-氯-4-氟苯胺的过程中产生3,4-二氟苯胺和4-氯-3-氟苯胺作为附属产物。由于这些附属产物具有基因毒性,因此在吉非替尼的生产过程中需要控制这些基因毒性杂质的含量。
王珊珊等人建立了液相色谱-串联质谱法测定吉非替尼中基因毒性杂质的含量。方法具体为:采用Accucore C18色谱柱(2.1 mm×100 mm,2.6μm),以0.1%甲酸水溶液-0.1%甲酸乙腈溶液(70:30)为流动相,流速0.3 mL·min-1,柱温30℃;采用ESI离子源正离子模式,多反应离子监测(MRM)模式下选择离子对m/z130.1→83.1(3,4-二氟苯胺)和m/z146.0→111.0(4-氯-3-氟苯胺)进行测定。
基因毒性杂质3,4-二氟苯胺和4-氯-3-氟苯胺的线性浓度范围为124 ng·mL-1,且线性关系良好(r=0.999 4、r=0.999 2);检测限为0.2 mg·kg-1,定量限为0.5 mg·kg-1;3,4-二氟苯胺和4-氯-3-氟苯胺的精密度试验的RSD(n=6)分别为2.3%和2.0%;杂质3,4-二氟苯胺低、中、高浓度的加标回收率(n=3)分别为99.4%、97.0%、105.5%,RSD=8.2%、5.1%、1.7%;杂质4-氯-3-氟苯胺低、中、高浓度的加标回收率(n=3)分别为102.0%、98.6%、106.0%,RSD分别为3.9%、7.9%、2.4%。经检测,3批吉非替尼供试品中3,4-二氟苯胺均未检出,批号2吉非替尼样品中4-氯-3-氟苯胺有少量检出,但低于样品的定量限(<0.5 mg·kg-1)。该方法操作简便,结果可靠,经方法学验证,可用于吉非替尼药物中基因毒性杂质3,4-二氟苯胺和4-氯-3-氟苯胺含量的同时测定。实验方法具体为:
(1)色谱条件:
采用色谱柱Accucore C18(2.1 mm× 100 mm,2.6 μm;填料:十八烷基硅烷键合硅胶),以0.1%甲酸水溶液-0.1%甲酸乙腈溶液(70∶30)为流动相,流速0.3 mL·min-1,柱温30 ℃,进样体积2 μL。
(2)质谱条件:
采用ESI离子源正离子模式,多反应离子监测(MRM)模式下选择离子对m/z 130.1→83.1(3,4-二氟苯胺)和m/z 146.0→111.0(4-氯-3-氟苯胺);解簇电压90 V(3,4-二氟苯胺)、76 V(4- 氯-3-氟苯胺);解离能量32 V(3,4-二氟苯胺)、26 V(4-氯-3-氟苯胺);入口电压10 V;出口电压 12 V;碰撞气压力Medium;气帘气压力1.03 MPa;雾化压力0.38 MPa;辅助加热气压力0.38 MPa;离子喷雾电压5.5 kV;离子源温度500 ℃。
(3)对照品溶液:
精密称取3,4-二氟苯胺、4- 氯-3-氟苯胺对照品各约10.01 mg,置100 mL量瓶中,加稀释剂乙腈-水(50:50)溶解并稀释至刻度,摇匀;精密量取1 mL,置100 mL量瓶中,用稀释剂乙腈-水(50:50)稀释至刻度,摇匀,配制成浓度为 1 μg·mL-1对照品储备液;精密量取1 mL,置10 mL 量瓶中,用稀释剂乙腈-水(50:50)稀释至刻度,摇匀;精密量取12 mL,置100 mL量瓶中,用稀释剂乙腈-水(50:50)稀释至刻度,摇匀,配成浓度为12 ng·mL-1的对照品溶液。
(4)供试品溶液 精密称取吉非替尼供试品约
20.03 mg,置10 mL量瓶中,用稀释剂乙腈-水(50:50) 振摇使溶解并稀释至刻度,取1 mL置2 mL离心管中,12 000 r·min-1离心10 min后取上清液,配制成浓度为2 mg·mL-1的吉非替尼供试品溶液。
参考文献:
[1]王珊珊,宁凡盛,王晓利等.HPLC-MS/MS法分析吉非替尼中痕量基因毒性杂质[J].药物分析杂志,2017,37(07):1309-1313.DOI:10.16155/j.0254-1793.2017.07.23
1
本文介绍了如何使用2,5-噻吩二羧酸来合成稀土金属有机配合物,旨在为相关研究人员提供参考依据。
背景:稀土金属有机配合物的发光性能具有强度高、波峰窄、颜色鲜艳以及耐候性好和不易被氧化等优点,日益受到人们的重视。有机羧酸极易与稀土离子生成稳定的稀土配合物,同时稀土离子具有较小的配位场稳定化能(一般只有4.18 kJ·mol-1),因而其配位方式多种多样,有较大和多变的配位数,使得稀土羧酸配合物的结构更加丰富多彩,确定其结构,研究其谱学性质,可为其应用提供理论基础。2,5-噻吩二羧酸(H2L)是一种共轭程度很高的杂环芳香羧酸,分子内含有较大的离域π键,且分子构型具有平面刚性结构。一般来说,这样的结构形成的稀土配合物由于刚性和共平面增加,可使外转移能量减少,从而有利于能量传递,提高稀土离子的发光效率。
应用:稀土金属有机配合物的合成
1. 合成轻稀土2,5-噻吩二羧酸-1,10-菲咯啉的三元配合物
张勇等人合成了几种新型的轻稀土2,5-噻吩二羧酸-1,10-菲咯啉的三元配合物。配合物的合成具体如下:
按Ln:H2L:Phen为2:3:2的比例称取1 mmol的Ln2O3,3 mmol的2,5-噻吩二羧酸和2 mmol的1,10-菲咯啉。稀土氧化物加浓盐酸加热溶解, 溶解后继续缓慢加热除去多余的盐酸得到带结晶水的稀土氯化物。待稀土氯化物冷却后用少量的去离子水将其溶解,在磁力搅拌器的不断搅动下,将2,5-噻吩二羧酸乙醇溶液逐滴滴加到稀土溶液中。调节反应液pH=5~6,有大量絮状沉淀生成,在60℃下反应回流50 min后,静置,抽滤,洗涤,烘干,封存。
2. 合成锌混合配体配合物
徐进等人合成锌混合配体配合物[Zn(thca)(phen)(H2O)](其中thca:2,5-噻吩二羧酸,phen:邻菲啰啉)。配合物的合成具体如下:
将10 mL、10 mmol/L的ZnCl2与10 mL、10 mmol/L的2,5-噻吩二羧酸(碱溶)混合,搅拌4h,将10 mL、10 mmol/L的邻菲啰啉(醇溶)加入混合溶液,持续搅拌2 h,然后用0.8 mol/L KOH溶液调节混合液pH值为6.42,经过滤 得到澄清溶液于小烧杯中,用保鲜膜封口,置于空气中,经过几周的溶剂挥发得到晶体。经 过Bruker Smart 1000 CCDC-X射线单晶衍射仪确定了以上所得晶体的结构为[Zn(thca) (phen)H2O]配合物。
3. 合成镧掺杂铕的2,5-噻吩二羧酸-1,10-菲咯啉四元异核配合物
李存雄等人合成了5种新型镧掺杂铕的2,5-噻吩二羧酸(H2L)、1,10-菲咯啉(Phen)四元异核配合物。配合物的合成具体如下:
按n(EuxLay)∶n(H2L)∶n(Phen)=2∶3∶2称取稀土氧化物(x∶y=0.90∶0.10、0.70∶0.30、0.50∶0.50、0.30∶0.70、 0.10∶0.90)、2, 5-噻吩二羧酸和1,10-菲咯啉。稀土氧化物加浓盐酸加热溶解,溶解后继续缓慢加热除去多余的盐酸得到带结晶水的稀土氯化物。
将2,5-噻吩二羧酸乙醇溶液和1,10-菲咯啉乙醇溶液用常压漏斗依次逐滴滴加到稀土溶液中。调节反应液pH=5~6,有大量絮状沉淀生成,6 0℃反应回流50min,静置、抽滤、洗涤、烘干、封存。
参考文献:
[1]罗署. 高纯噻吩-2,5-二羧酸的合成 [J]. 染料与染色, 2016, 53 (01): 41-42+47.
[2]徐进,姜美平,武光磊等. 配合物锌-2,5-噻吩二羧酸-邻菲啰啉的合成及其与DNA作用的研究 [J]. 沈阳化工大学学报, 2012, 26 (03): 193-198.
[3]张勇,廖莉玲,邹文静等. 轻稀土-2,5-噻吩二羧酸-1,10-菲咯啉三元配合物的合成、表征及谱学性质 [J]. 稀土, 2011, 32 (04): 8-13. DOI:10.16533/j.cnki.15-1099/tf.2011.04.007
[4]李存雄,张勇,廖莉玲等. 镧掺杂铕的2,5-噻吩二羧酸-1,10-菲咯啉四元异核配合物的合成、表征及谱学性质 [J]. 材料导报, 2011, 25 (04): 49-52.
显示全部本文介绍了如何使用2,5-噻吩二羧酸来合成稀土金属有机配合物,旨在为相关研究人员提供参考依据。
背景:稀土金属有机配合物的发光性能具有强度高、波峰窄、颜色鲜艳以及耐候性好和不易被氧化等优点,日益受到人们的重视。有机羧酸极易与稀土离子生成稳定的稀土配合物,同时稀土离子具有较小的配位场稳定化能(一般只有4.18 kJ·mol-1),因而其配位方式多种多样,有较大和多变的配位数,使得稀土羧酸配合物的结构更加丰富多彩,确定其结构,研究其谱学性质,可为其应用提供理论基础。2,5-噻吩二羧酸(H2L)是一种共轭程度很高的杂环芳香羧酸,分子内含有较大的离域π键,且分子构型具有平面刚性结构。一般来说,这样的结构形成的稀土配合物由于刚性和共平面增加,可使外转移能量减少,从而有利于能量传递,提高稀土离子的发光效率。
应用:稀土金属有机配合物的合成
1. 合成轻稀土2,5-噻吩二羧酸-1,10-菲咯啉的三元配合物
张勇等人合成了几种新型的轻稀土2,5-噻吩二羧酸-1,10-菲咯啉的三元配合物。配合物的合成具体如下:
按Ln:H2L:Phen为2:3:2的比例称取1 mmol的Ln2O3,3 mmol的2,5-噻吩二羧酸和2 mmol的1,10-菲咯啉。稀土氧化物加浓盐酸加热溶解, 溶解后继续缓慢加热除去多余的盐酸得到带结晶水的稀土氯化物。待稀土氯化物冷却后用少量的去离子水将其溶解,在磁力搅拌器的不断搅动下,将2,5-噻吩二羧酸乙醇溶液逐滴滴加到稀土溶液中。调节反应液pH=5~6,有大量絮状沉淀生成,在60℃下反应回流50 min后,静置,抽滤,洗涤,烘干,封存。
2. 合成锌混合配体配合物
徐进等人合成锌混合配体配合物[Zn(thca)(phen)(H2O)](其中thca:2,5-噻吩二羧酸,phen:邻菲啰啉)。配合物的合成具体如下:
将10 mL、10 mmol/L的ZnCl2与10 mL、10 mmol/L的2,5-噻吩二羧酸(碱溶)混合,搅拌4h,将10 mL、10 mmol/L的邻菲啰啉(醇溶)加入混合溶液,持续搅拌2 h,然后用0.8 mol/L KOH溶液调节混合液pH值为6.42,经过滤 得到澄清溶液于小烧杯中,用保鲜膜封口,置于空气中,经过几周的溶剂挥发得到晶体。经 过Bruker Smart 1000 CCDC-X射线单晶衍射仪确定了以上所得晶体的结构为[Zn(thca) (phen)H2O]配合物。
3. 合成镧掺杂铕的2,5-噻吩二羧酸-1,10-菲咯啉四元异核配合物
李存雄等人合成了5种新型镧掺杂铕的2,5-噻吩二羧酸(H2L)、1,10-菲咯啉(Phen)四元异核配合物。配合物的合成具体如下:
按n(EuxLay)∶n(H2L)∶n(Phen)=2∶3∶2称取稀土氧化物(x∶y=0.90∶0.10、0.70∶0.30、0.50∶0.50、0.30∶0.70、 0.10∶0.90)、2, 5-噻吩二羧酸和1,10-菲咯啉。稀土氧化物加浓盐酸加热溶解,溶解后继续缓慢加热除去多余的盐酸得到带结晶水的稀土氯化物。
将2,5-噻吩二羧酸乙醇溶液和1,10-菲咯啉乙醇溶液用常压漏斗依次逐滴滴加到稀土溶液中。调节反应液pH=5~6,有大量絮状沉淀生成,6 0℃反应回流50min,静置、抽滤、洗涤、烘干、封存。
参考文献:
[1]罗署. 高纯噻吩-2,5-二羧酸的合成 [J]. 染料与染色, 2016, 53 (01): 41-42+47.
[2]徐进,姜美平,武光磊等. 配合物锌-2,5-噻吩二羧酸-邻菲啰啉的合成及其与DNA作用的研究 [J]. 沈阳化工大学学报, 2012, 26 (03): 193-198.
[3]张勇,廖莉玲,邹文静等. 轻稀土-2,5-噻吩二羧酸-1,10-菲咯啉三元配合物的合成、表征及谱学性质 [J]. 稀土, 2011, 32 (04): 8-13. DOI:10.16533/j.cnki.15-1099/tf.2011.04.007
[4]李存雄,张勇,廖莉玲等. 镧掺杂铕的2,5-噻吩二羧酸-1,10-菲咯啉四元异核配合物的合成、表征及谱学性质 [J]. 材料导报, 2011, 25 (04): 49-52.
1
本文将介绍测定2, 4-二氨基苯氧基乙醇盐酸盐的方法,通过深入了解2, 4-二氨基苯氧基乙醇盐酸盐的检测方法,我们可以更好地理解该物质的应用与意义。
背景:《化妆品安全技术规范》(2015年版)规定,化妆品准用染料2, 4-二氨基苯氧基乙醇盐酸盐在氧化型染发产品中最大允许浓度不超过质量分数2.0%。否则,长期使用染料含量超标的氧化型染发产品可引发过敏、皮肤损害及眼损害等,严重的甚至会致畸、致癌、致突变。因此,对2, 4-二氨基苯氧基乙醇盐酸盐含量的检测对染发剂的质量监控和保障人体健康有重要意义。
1. 合成:
采用2,4-二硝基氯苯和乙二醇为原料,在NaOH 催化下直接通过Williamson 反应合成2,4-二硝基苯氧基乙醇;通过催化氢化方法在低压下由2,4-二硝基苯氧基乙醇可制备2,4-二氨基苯氧基乙醇盐酸盐。
2. 测定:
2.1 张小媚等人建立了高效液相色谱法测定化妆品中2,4-二氨基苯氧基乙醇盐酸盐含量不确定度评定的数学模型,分析了检测 过程中不确定度的来源,对不确定度进行了评定。结果显示该物质含量的相对合成标准不确定度为0.0027%,相对扩展不确定度为0.005 4%。该评定为验证测量结果的可信程度提供依据。其中测定方法如下:
(1)液相色谱条件:
Discovery RP Amide C16色谱柱(250 mm×4.6 mm×5 μm);流动相:甲醇和磷酸盐混合溶液(pH为6),体积比为3∶97;流速:1.0 mL/min;检测波长:280 nm;柱温:25 ℃;进样量:5 μL。
(2)测定方法:
标准溶液配制:精密称取2, 4-二氨基苯氧基乙醇盐酸盐对照品0.1 g,置10 mL量瓶中,以 2 g/L亚硫酸氢钠水溶液和无水乙醇(体积比1∶1) 的混合溶液溶解并定容至刻度,制成质量浓度为 10 g/L的标准储备溶液。取标准储备溶液5 mL于 100 mL量瓶中,用无水乙醇稀释至刻度,配制成质量浓度为500 mg/L的标准溶液;取500 mg/ L标 准溶液适量,用无水乙醇稀释,配制成质量浓度为10、25、50、100、250 mg/L的标准系列溶液 S1~S5,临用现配。
(3)样品处理:
称取样品0.5 g(精确到0.001 g)于10 mL量瓶中,加无水乙醇和水(体积比1∶1)的混合溶液至刻度,涡旋1 min,冰浴超声提取 15 min。取适量离心(5 000 r/min)5 min,取上清液经0.45 μm微孔滤膜过滤,滤液作为待测溶液,并尽快测定。
(4)测定:
取2, 4-二氨基苯氧基乙醇盐酸盐系列标准溶液S1~S5待测液5 μL进样,色谱分析测定,以待测液浓度为横坐标、以其对应的峰面积为纵坐标,做标准曲线。取样品待测液5 μL进样 以出峰时间及DAD光谱图定性,峰面积定量,标准曲线法计算样品中2, 4-二氨基苯氧基乙醇盐酸盐的含量。
2.2 王莉等人建立了同时测定2,4-二氨基苯氧基乙醇盐酸盐和2-氨基-4-羟乙氨基茴香醚硫酸盐的高效液相色谱方法。样品经用甲醇-5 mmol/L乙酸铵溶液(含亚硫酸氢钠2 g/L)(1∶1)作为流动相,采用C18色谱柱进行分离,以甲醇-5 mmol/L乙酸铵溶液 (13∶87)为流动相进行洗脱,检测波长240 nm,流速1.0 m L/min,柱温30℃,结果表明两种染发剂色谱峰分离良好,两种染发剂标准曲线线性系数为0.999,低、中、高三种水平的加标回收率为98.6%~105.5%,相对标准偏差为0.9%~2.3%。该方法简便快速,可用于两种染料的同时测定。
参考文献:
[1]王莉,曾浩洋. HPLC同时测定2,4-二氨基苯氧基乙醇盐酸盐和2-氨基-4-羟乙氨基茴香醚硫酸盐 [J]. 广州化工, 2023, 51 (09): 91-93.
[2]张小媚,周智明,陈桂琴等. 高效液相色谱法测定化妆品中2,4-二氨基苯氧基乙醇盐酸盐含量的不确定度 [J]. 香料香精化妆品, 2022, (03): 85-88.
[3]朱翼鸣, 2,4-二氨基苯氧基乙醇盐酸盐. 浙江省, 浙江鼎龙科技有限公司, 2015-06-03.
显示全部本文将介绍测定2, 4-二氨基苯氧基乙醇盐酸盐的方法,通过深入了解2, 4-二氨基苯氧基乙醇盐酸盐的检测方法,我们可以更好地理解该物质的应用与意义。
背景:《化妆品安全技术规范》(2015年版)规定,化妆品准用染料2, 4-二氨基苯氧基乙醇盐酸盐在氧化型染发产品中最大允许浓度不超过质量分数2.0%。否则,长期使用染料含量超标的氧化型染发产品可引发过敏、皮肤损害及眼损害等,严重的甚至会致畸、致癌、致突变。因此,对2, 4-二氨基苯氧基乙醇盐酸盐含量的检测对染发剂的质量监控和保障人体健康有重要意义。
1. 合成:
采用2,4-二硝基氯苯和乙二醇为原料,在NaOH 催化下直接通过Williamson 反应合成2,4-二硝基苯氧基乙醇;通过催化氢化方法在低压下由2,4-二硝基苯氧基乙醇可制备2,4-二氨基苯氧基乙醇盐酸盐。
2. 测定:
2.1 张小媚等人建立了高效液相色谱法测定化妆品中2,4-二氨基苯氧基乙醇盐酸盐含量不确定度评定的数学模型,分析了检测 过程中不确定度的来源,对不确定度进行了评定。结果显示该物质含量的相对合成标准不确定度为0.0027%,相对扩展不确定度为0.005 4%。该评定为验证测量结果的可信程度提供依据。其中测定方法如下:
(1)液相色谱条件:
Discovery RP Amide C16色谱柱(250 mm×4.6 mm×5 μm);流动相:甲醇和磷酸盐混合溶液(pH为6),体积比为3∶97;流速:1.0 mL/min;检测波长:280 nm;柱温:25 ℃;进样量:5 μL。
(2)测定方法:
标准溶液配制:精密称取2, 4-二氨基苯氧基乙醇盐酸盐对照品0.1 g,置10 mL量瓶中,以 2 g/L亚硫酸氢钠水溶液和无水乙醇(体积比1∶1) 的混合溶液溶解并定容至刻度,制成质量浓度为 10 g/L的标准储备溶液。取标准储备溶液5 mL于 100 mL量瓶中,用无水乙醇稀释至刻度,配制成质量浓度为500 mg/L的标准溶液;取500 mg/ L标 准溶液适量,用无水乙醇稀释,配制成质量浓度为10、25、50、100、250 mg/L的标准系列溶液 S1~S5,临用现配。
(3)样品处理:
称取样品0.5 g(精确到0.001 g)于10 mL量瓶中,加无水乙醇和水(体积比1∶1)的混合溶液至刻度,涡旋1 min,冰浴超声提取 15 min。取适量离心(5 000 r/min)5 min,取上清液经0.45 μm微孔滤膜过滤,滤液作为待测溶液,并尽快测定。
(4)测定:
取2, 4-二氨基苯氧基乙醇盐酸盐系列标准溶液S1~S5待测液5 μL进样,色谱分析测定,以待测液浓度为横坐标、以其对应的峰面积为纵坐标,做标准曲线。取样品待测液5 μL进样 以出峰时间及DAD光谱图定性,峰面积定量,标准曲线法计算样品中2, 4-二氨基苯氧基乙醇盐酸盐的含量。
2.2 王莉等人建立了同时测定2,4-二氨基苯氧基乙醇盐酸盐和2-氨基-4-羟乙氨基茴香醚硫酸盐的高效液相色谱方法。样品经用甲醇-5 mmol/L乙酸铵溶液(含亚硫酸氢钠2 g/L)(1∶1)作为流动相,采用C18色谱柱进行分离,以甲醇-5 mmol/L乙酸铵溶液 (13∶87)为流动相进行洗脱,检测波长240 nm,流速1.0 m L/min,柱温30℃,结果表明两种染发剂色谱峰分离良好,两种染发剂标准曲线线性系数为0.999,低、中、高三种水平的加标回收率为98.6%~105.5%,相对标准偏差为0.9%~2.3%。该方法简便快速,可用于两种染料的同时测定。
参考文献:
[1]王莉,曾浩洋. HPLC同时测定2,4-二氨基苯氧基乙醇盐酸盐和2-氨基-4-羟乙氨基茴香醚硫酸盐 [J]. 广州化工, 2023, 51 (09): 91-93.
[2]张小媚,周智明,陈桂琴等. 高效液相色谱法测定化妆品中2,4-二氨基苯氧基乙醇盐酸盐含量的不确定度 [J]. 香料香精化妆品, 2022, (03): 85-88.
[3]朱翼鸣, 2,4-二氨基苯氧基乙醇盐酸盐. 浙江省, 浙江鼎龙科技有限公司, 2015-06-03.
1
对氨基苯磺酸钠是一种常见的化合物,其检测方法具有重要意义。本文将介绍其检测方法,为准确快速地检测对氨基苯磺酸钠提供参考依据。
背景:对氨基苯磺酸钠,分子式为C6H6NO3SNa,纯品为有光泽的白色结晶,易溶于水,不溶于一般的有机溶剂,水溶液呈中性,遇含钙的物质会产生沉淀。对氨基苯磺酸钠的合成路线见图。
检测:
目前,对氨基苯磺酸钠的研究多是将其作为食品医药中间体或杀菌剂,关于对氨基苯磺酸钠的检测方法较少,主要是磺酸盐和磺酸酯类的分析检测方法,检测方法有紫外分光光度法、气相色谱法、气质联用法、高效液相色谱法、液质联用法。
(1)紫外分光光度法
李占彬等建立了检测地沟油中十二烷基苯磺酸钠的方法,该法通过十二烷基苯磺酸钠与亚甲蓝生成显色物质,再用紫外分光光度计进行测定。结果表明, 检出限为0.1 μg/kg,十二烷基苯磺酸钠在0.5-4.0 mg/L范围内呈良好线性关系,不同浓度的平均回收率为94%-96%,相对标准偏差为2.5%-4.6%。紫外分光光度操作简单,设备便宜,但灵敏度较低,抗干扰较弱,达不到药品中痕量杂质检测要求。
(2)气相色谱法
张志强等采用气相色谱法测定秋梨膏中的环己基氨基磺酸钠含量,实验结果表明,环己基氨基磺酸钠最低检出限为2 mg/kg,在0.05-1.00 g/L内有良好 的线性关系,加标回收率为90.0%-97.5%(n=6),相对标准偏差3.31%-9.05%。 气相色谱适应易于气化、热稳定性好、沸点较低的样品分析,不适宜于挥发性差、离子型样品分析,但对氨基苯磺酸钠属于离子型不易挥发样品。
(3)气质联用法
曹卫等建立了检测甲磺酸萘莫司他中基因毒性杂质甲磺酸甲酯的气质联用法,使用二氯甲烷进行提取,测得甲磺酸甲酯的最低检测限为5 ng/mL,最小定量限为14.8 ng/mL,在0.01-0.4 μg/mL的浓度范围内呈线性关系,平均回收率为90.2%(RSD=1.5%)。
(4)高效液相色谱法
谢卫锋等利用高效液相色谱法检测托吡司特中的基因毒性杂质对甲苯磺酸乙酯,该方法的检测限为0.0168μg/mL,对甲苯磺酸乙酯在质量浓度为 0.051-0.204 μg/mL范围内与峰面积呈良好线性关系,平均回收率为98.2%。
(5)液质联用法
王康林等利用高效液相色谱-质谱联用法测定伊马替尼中基因毒性杂质甲 磺酸乙酯,该方法通过加入过量的衍生化试剂2-巯基吡啶与甲磺酸乙酯衍生化, 再用色谱检测。结果表明甲磺酸乙酯浓度在4.69 ng/mL-14.06 ng/mL范围内呈良好线性关系,检测限为1.4 ng/mL,定量限为4.7 ng/mL,平均回收率为98.0% (RSD=1.6 %)
参考文献:
[1]起燕江,曾涛,朱梓良. HPLC法测定对氨基苯磺酸钠中的同分异构体 [J]. 药物评价研究, 2022, 45 (03): 504-508.
[2]叶思东. 原料药中基因毒性杂质甲醛及对氨基苯磺酸钠的检测[D]. 东华大学, 2019. DOI:10.27012/d.cnki.gdhuu.2019.000370
显示全部对氨基苯磺酸钠是一种常见的化合物,其检测方法具有重要意义。本文将介绍其检测方法,为准确快速地检测对氨基苯磺酸钠提供参考依据。
背景:对氨基苯磺酸钠,分子式为C6H6NO3SNa,纯品为有光泽的白色结晶,易溶于水,不溶于一般的有机溶剂,水溶液呈中性,遇含钙的物质会产生沉淀。对氨基苯磺酸钠的合成路线见图。
检测:
目前,对氨基苯磺酸钠的研究多是将其作为食品医药中间体或杀菌剂,关于对氨基苯磺酸钠的检测方法较少,主要是磺酸盐和磺酸酯类的分析检测方法,检测方法有紫外分光光度法、气相色谱法、气质联用法、高效液相色谱法、液质联用法。
(1)紫外分光光度法
李占彬等建立了检测地沟油中十二烷基苯磺酸钠的方法,该法通过十二烷基苯磺酸钠与亚甲蓝生成显色物质,再用紫外分光光度计进行测定。结果表明, 检出限为0.1 μg/kg,十二烷基苯磺酸钠在0.5-4.0 mg/L范围内呈良好线性关系,不同浓度的平均回收率为94%-96%,相对标准偏差为2.5%-4.6%。紫外分光光度操作简单,设备便宜,但灵敏度较低,抗干扰较弱,达不到药品中痕量杂质检测要求。
(2)气相色谱法
张志强等采用气相色谱法测定秋梨膏中的环己基氨基磺酸钠含量,实验结果表明,环己基氨基磺酸钠最低检出限为2 mg/kg,在0.05-1.00 g/L内有良好 的线性关系,加标回收率为90.0%-97.5%(n=6),相对标准偏差3.31%-9.05%。 气相色谱适应易于气化、热稳定性好、沸点较低的样品分析,不适宜于挥发性差、离子型样品分析,但对氨基苯磺酸钠属于离子型不易挥发样品。
(3)气质联用法
曹卫等建立了检测甲磺酸萘莫司他中基因毒性杂质甲磺酸甲酯的气质联用法,使用二氯甲烷进行提取,测得甲磺酸甲酯的最低检测限为5 ng/mL,最小定量限为14.8 ng/mL,在0.01-0.4 μg/mL的浓度范围内呈线性关系,平均回收率为90.2%(RSD=1.5%)。
(4)高效液相色谱法
谢卫锋等利用高效液相色谱法检测托吡司特中的基因毒性杂质对甲苯磺酸乙酯,该方法的检测限为0.0168μg/mL,对甲苯磺酸乙酯在质量浓度为 0.051-0.204 μg/mL范围内与峰面积呈良好线性关系,平均回收率为98.2%。
(5)液质联用法
王康林等利用高效液相色谱-质谱联用法测定伊马替尼中基因毒性杂质甲 磺酸乙酯,该方法通过加入过量的衍生化试剂2-巯基吡啶与甲磺酸乙酯衍生化, 再用色谱检测。结果表明甲磺酸乙酯浓度在4.69 ng/mL-14.06 ng/mL范围内呈良好线性关系,检测限为1.4 ng/mL,定量限为4.7 ng/mL,平均回收率为98.0% (RSD=1.6 %)
参考文献:
[1]起燕江,曾涛,朱梓良. HPLC法测定对氨基苯磺酸钠中的同分异构体 [J]. 药物评价研究, 2022, 45 (03): 504-508.
[2]叶思东. 原料药中基因毒性杂质甲醛及对氨基苯磺酸钠的检测[D]. 东华大学, 2019. DOI:10.27012/d.cnki.gdhuu.2019.000370
1
本文将介绍测定甲氧氯普胺的含量的方法,通过深入了解甲氧氯普胺的检测方法,我们可以更好地理解该物质的应用与意义。
背景:甲氧氯普胺为一种常用的镇吐药品,可用于因脑部肿瘤手术、肿瘤的放疗及化疗、脑外伤后遗症、急性颅脑损伤以及药物所引起的呕吐。对于胃胀 气性消化不良、食欲不振、嗳气、恶心、呕吐也有较好的疗效。
测定:
1. 气相色谱分析方法
赵磊等人建立甲氧氯普胺的气相色谱分析方法。方法为:采用毛细管色谱柱;流动相:氮气;检测器:μECD;流速:1.0 mL/min;进样口温度:300℃;柱温:280℃;检测器温度:300℃。甲氧氯普胺在0.25~ 50 μg/mL范围内与峰面积具有良好的线性关系;加样回收率均符合要求。该方法简便快捷,准确、重 性好,灵敏度高,可用于甲氧氯普胺的含量检测方法。
2. 流动注射化学发光分析法
(1)熊海涛等人根据在酸性条件下,溴化钠对NaIO4-H2O2-甲氧氯普胺弱化学发光具有增敏作用,结合流动注射技术建立了一种测定甲氧氯普胺的新方法。在优化实验条件下,相对化学发光强度与甲氧氯普胺的质量浓度在 1.00×10- 9 ~1.20×10-6 g/m L范围内呈良好的线性关系,检出限为3.5×10-10 g/m L,相对标准偏差(ρ= 1.00×10- 7 g/mL,n=11)为2.5%。该方法已成功用于片剂及加标尿样中甲氧氯普胺含量的测定,回收率为 96%~100%,与标准方法相对照,测定结果基本一致。实验方法为:
流动注射化学发光仪流路图如图所示。首先,传送试样溶液流过并使其完全充满管路,然后传送NaIO4、H2O2、H3PO4至管路,此时试样和体系在V点混合发生化学发光反应,NaIO4 -H2O2 混合液流经光电倍增管时体系的发光强度输出为I,同时测定试剂空白的发光强度I0,以相对发光强度ΔI (I-I0)对甲氧氯普胺标准溶液的浓度作图,从而进行定量分析。
(2)吴志皓等人基于在甲醛的作用下,高锰酸钾对甲氧氯普胺的氧化作用而产生化学发光的现象,建立了一种新的用流动注射-化学发光法测定甲氧氯普胺含量的方法。该方法测定甲氧氯普胺的线性范围为0.2100 mg/L,检出限为0.1 mg/L.对于8 mg/L的甲氧氯普胺标准溶液连续11次测定的相对偏差为1.2%.该方法可用于对制药废水、片剂和针剂中甲氧氯普胺含量的测定。实验方法为:
泵载流速均为2.3 mL/min,管径为0.8 mm,阀池距为2 cm,光电倍增管高压为900 V.待测MCPM试液与甲醛溶液经三通混合后,由六通阀定量注射(75 μL)入经三通混合的KMnO4溶液与HCl的混合液中,反应产生化学发光,光电倍增管检测发光信号,相对发光信号与MCPM浓度成正比,由计算机记录发光强度、时间等信号。
3. 流动注射计时电流法
彭亚鸽基于多壁碳纳米管修饰印刷碳电极对甲氧氯普胺的电催化作用,建立了测定甲氧氯普胺的流动注射计时电流法。与印刷碳电极相比,多壁碳纳米管修饰印刷碳电极显著降低了甲氧氯普胺的氧化峰电位,提高了氧化峰电流.测定甲氧氯普胺的线性范围为8.04×10-5~1.0×10-3 mol/L,检出限为5.0×10-5 mol/L(S/N=3);1.0×10-4 mol/L 的甲氧氯普胺测定的RSD为3.0%(n=9).方法已成功应用于药片中甲氧氯普胺含量的测定。实验方法为:
流动注射流路如图所示。将印刷碳电极或 MWCNT修饰印刷碳电极、铂丝对电极和Ag/AgCl 参比电极插入流通池,电极末端与CHI660B电化学 工作站相连,然后将MCPM以1 mL/min的流速经管道b通入载流(NaAc-HAc缓冲溶液,pH=5.00),同时施加1.3 V的恒电位。基线稳定后,通过采样阀注入甲氧氯普胺标准溶液或样品,记录相应信号。
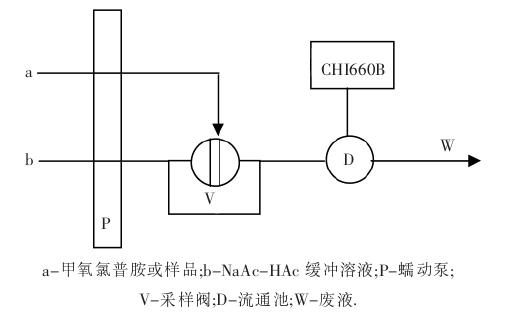
参考文献:
[1]赵磊,于丹,赵宾等. 甲氧氯普胺片的气相色谱分析方法 [J]. 中国合理用药探索, 2017, 14 (09): 71-73.
[2]熊海涛,唐志华,聂峰等. 流动注射化学发光分析法测定片剂甲氧氯普胺 [J]. 分析测试学报, 2013, 32 (02): 244-248.
[3]彭亚鸽,谭颖,李丛丛. 流动注射计时电流法检测甲氧氯普胺 [J]. 宁夏工程技术, 2012, 11 (04): 341-344.
[4]吴志皓,李桂敏,王金中等. 高锰酸钾-甲醛化学发光体系测定甲氧氯普胺 [J]. 化学研究, 2005, (03): 87-89.
显示全部本文将介绍测定甲氧氯普胺的含量的方法,通过深入了解甲氧氯普胺的检测方法,我们可以更好地理解该物质的应用与意义。
背景:甲氧氯普胺为一种常用的镇吐药品,可用于因脑部肿瘤手术、肿瘤的放疗及化疗、脑外伤后遗症、急性颅脑损伤以及药物所引起的呕吐。对于胃胀 气性消化不良、食欲不振、嗳气、恶心、呕吐也有较好的疗效。
测定:
1. 气相色谱分析方法
赵磊等人建立甲氧氯普胺的气相色谱分析方法。方法为:采用毛细管色谱柱;流动相:氮气;检测器:μECD;流速:1.0 mL/min;进样口温度:300℃;柱温:280℃;检测器温度:300℃。甲氧氯普胺在0.25~ 50 μg/mL范围内与峰面积具有良好的线性关系;加样回收率均符合要求。该方法简便快捷,准确、重 性好,灵敏度高,可用于甲氧氯普胺的含量检测方法。
2. 流动注射化学发光分析法
(1)熊海涛等人根据在酸性条件下,溴化钠对NaIO4-H2O2-甲氧氯普胺弱化学发光具有增敏作用,结合流动注射技术建立了一种测定甲氧氯普胺的新方法。在优化实验条件下,相对化学发光强度与甲氧氯普胺的质量浓度在 1.00×10- 9 ~1.20×10-6 g/m L范围内呈良好的线性关系,检出限为3.5×10-10 g/m L,相对标准偏差(ρ= 1.00×10- 7 g/mL,n=11)为2.5%。该方法已成功用于片剂及加标尿样中甲氧氯普胺含量的测定,回收率为 96%~100%,与标准方法相对照,测定结果基本一致。实验方法为:
流动注射化学发光仪流路图如图所示。首先,传送试样溶液流过并使其完全充满管路,然后传送NaIO4、H2O2、H3PO4至管路,此时试样和体系在V点混合发生化学发光反应,NaIO4 -H2O2 混合液流经光电倍增管时体系的发光强度输出为I,同时测定试剂空白的发光强度I0,以相对发光强度ΔI (I-I0)对甲氧氯普胺标准溶液的浓度作图,从而进行定量分析。
(2)吴志皓等人基于在甲醛的作用下,高锰酸钾对甲氧氯普胺的氧化作用而产生化学发光的现象,建立了一种新的用流动注射-化学发光法测定甲氧氯普胺含量的方法。该方法测定甲氧氯普胺的线性范围为0.2100 mg/L,检出限为0.1 mg/L.对于8 mg/L的甲氧氯普胺标准溶液连续11次测定的相对偏差为1.2%.该方法可用于对制药废水、片剂和针剂中甲氧氯普胺含量的测定。实验方法为:
泵载流速均为2.3 mL/min,管径为0.8 mm,阀池距为2 cm,光电倍增管高压为900 V.待测MCPM试液与甲醛溶液经三通混合后,由六通阀定量注射(75 μL)入经三通混合的KMnO4溶液与HCl的混合液中,反应产生化学发光,光电倍增管检测发光信号,相对发光信号与MCPM浓度成正比,由计算机记录发光强度、时间等信号。
3. 流动注射计时电流法
彭亚鸽基于多壁碳纳米管修饰印刷碳电极对甲氧氯普胺的电催化作用,建立了测定甲氧氯普胺的流动注射计时电流法。与印刷碳电极相比,多壁碳纳米管修饰印刷碳电极显著降低了甲氧氯普胺的氧化峰电位,提高了氧化峰电流.测定甲氧氯普胺的线性范围为8.04×10-5~1.0×10-3 mol/L,检出限为5.0×10-5 mol/L(S/N=3);1.0×10-4 mol/L 的甲氧氯普胺测定的RSD为3.0%(n=9).方法已成功应用于药片中甲氧氯普胺含量的测定。实验方法为:
流动注射流路如图所示。将印刷碳电极或 MWCNT修饰印刷碳电极、铂丝对电极和Ag/AgCl 参比电极插入流通池,电极末端与CHI660B电化学 工作站相连,然后将MCPM以1 mL/min的流速经管道b通入载流(NaAc-HAc缓冲溶液,pH=5.00),同时施加1.3 V的恒电位。基线稳定后,通过采样阀注入甲氧氯普胺标准溶液或样品,记录相应信号。
参考文献:
[1]赵磊,于丹,赵宾等. 甲氧氯普胺片的气相色谱分析方法 [J]. 中国合理用药探索, 2017, 14 (09): 71-73.
[2]熊海涛,唐志华,聂峰等. 流动注射化学发光分析法测定片剂甲氧氯普胺 [J]. 分析测试学报, 2013, 32 (02): 244-248.
[3]彭亚鸽,谭颖,李丛丛. 流动注射计时电流法检测甲氧氯普胺 [J]. 宁夏工程技术, 2012, 11 (04): 341-344.
[4]吴志皓,李桂敏,王金中等. 高锰酸钾-甲醛化学发光体系测定甲氧氯普胺 [J]. 化学研究, 2005, (03): 87-89.

|

|
 关注盖德视界
关注盖德视界
 添加小助手
添加小助手




















